ClockstaR
1.0.0

Sebastian Duchene, Martyna Molak und Simon YW Ho.
Labor für Molekulare Ökologie, Evolution und Phylogenetik (MEEP).
Fakultät für Biowissenschaften
Universität Sydney
10. Juni 2015
partition_data_partitionfinder('drag fasta file with concatenated data here', 'drag partition finder output here')
optim.trees.interactive(folder.parts = 'path to your folder with fasta files and tree topology here')
Implementieren Sie die Optimierung von Baumabständen mithilfe der Ableitung des BSD-Abstands
Implementieren Sie eine parallele Version für die Topologieentfernung
Schreiben Sie ein Tutorial für Topologie-Distanz-Clustering
Integrieren Sie den Modellgenerator für Modelltests
Integrieren Sie RaxML zur Maximum-Likelihood-Optimierung von Zweiglängen und Topologien
Die Schätzung evolutionärer Zeitskalen anhand von Multigen-Datensätzen ist eine häufige Aufgabe in phylogenetischen Studien. Multigen-Datensätze können nach Gen, Codon-Position oder beidem unterteilt werden. In diesem Tutorial bezeichnen wir „Datenteilmengen“ als einzelne Gene oder jede Untereinheit des Multigen-Datensatzes. Der Begriff „Partitionen“ bezieht sich auf eine Gruppe von Datenteilmengen.
Obwohl die Datenteilmengen mit einem einzigen Relaxed-Clock-Modell verkettet und analysiert werden können, können sich die Muster der Ratenvariation zwischen den Abstammungslinien zwischen den Datenteilmengen unterscheiden, selbst wenn ihre Baumtopologien identisch sind. Beispielsweise kann sich die Geschwindigkeitsvariation zwischen den Abstammungslinien bei mitochondrialen Genen von der bei Kerngenen unterscheiden. Daher können unterschiedliche Relaxed-Clock-Modelle unterschiedlichen Datenteilmengen zugeordnet werden, um die Schätzungen evolutionärer Zeitskalen und die statistische Passung zu verbessern (siehe Duchene und Ho., 2014a).
Es gibt zahlreiche Möglichkeiten, Multigen-Datensätze zu partitionieren. Ein gängiger Ansatz zum Vergleich von Partitionierungsschemata ist die Verwendung von Bayes-Faktoren oder wahrscheinlichkeitsbasierten Kriterien für die Modellanpassung. In den meisten Fällen ist es jedoch nicht möglich, alle möglichen Partitionierungsschemata zu testen, insbesondere mit rechenintensiven Methoden zur Berechnung von Bayes-Faktoren.
ClockstaR schätzt die phylogenetischen Zweiglängen jeder Datenteilmenge. Der Branch-Score-Abstand, bekannt als sBSDmin, wird für jedes Baumpaar als Maß für den Unterschied in ihren Mustern der Variationsrate zwischen den Abstammungslinien berechnet. Diese Abstände werden verwendet, um die beste Partitionierungsstrategie mithilfe der GAP-Statistik mit dem PAM-Clustering-Algorithmus abzuleiten, wie er im Paketcluster implementiert ist (Maechler et al., 2012) (Einzelheiten zur sBSDmin-Metrik finden Sie bei Duchene et al., 2014b). .
ClockstaR ist ein R-Paket für phylogenetische molekulare Uhrenanalysen von Multi-Gen-Datensätzen. Es verwendet die Muster der Variation der Abstammungsraten für die verschiedenen Gene, um die Taktaufteilungsstrategie auszuwählen. Die Methode verwendet eine phylogenetische Baumdistanzmetrik und einen unbeaufsichtigten Algorithmus für maschinelles Lernen, um die optimale Anzahl von Uhrenpartitionen zu ermitteln und zu ermitteln, welche Gene unter den einzelnen Partitionen analysiert werden sollten. Die in ClocsktaR ausgewählte Partitionierungsstrategie kann für die anschließende Analyse der molekularen Uhr mit Programmen wie BEAST, MrBayes, PhyloBayes und anderen verwendet werden.
Bitte folgen Sie diesem Link für die Originalveröffentlichung.
ClockstaR erfordert eine R-Installation. Es erfordert auch einige R-Abhängigkeiten, die über R abgerufen werden können, wie unten erläutert.
Bitte senden Sie Ihre Wünsche oder Fragen an Sebastian Duchene (sebastian.duchene[at]sydney.edu.au). Weitere Software und Ressourcen finden Sie im Molecular Ecology, Evolution, and Phylogenetics Laboratory der University of Sydney.
Laden Sie dieses Repository als ZIP-Datei herunter und entpacken Sie es. Die folgenden Anweisungen verwenden den Ordner „clockstar_example_data“, der einige Fasta-Dateien und einen phylogenetischen Baum im Newick-Format enthält. Öffnen Sie eine dieser Dateien in einem Texteditor, z. B. Text Wrangler. Diese Daten wurden unter vier Mustern der Variation der Evolutionsrate simuliert. Beachten Sie, dass der Baum die Baumtopologie für alle Gene oder Datenpartitionen ist. Um ClockstaR auszuführen, formatieren Sie bitte Ihre Daten ähnlich den Beispieldaten in clockstar_example_data.
ClockstaR kann direkt von GitHub installiert werden. Hierzu ist das Devtools-Paket erforderlich. Geben Sie an der R-Eingabeaufforderung den folgenden Code ein, um alle erforderlichen Tools zu installieren (beachten Sie, dass Sie eine Internetverbindung benötigen, um die Pakete direkt herunterzuladen):.
install . packages ( " devtools " )
library (devtools)
install_github ( ' ClockstaR ' , ' sebastianduchene ' )Laden Sie nach dem Herunterladen und Installieren ClockstaR mit der Funktionsbibliothek .
library (ClockstaR2)Um ein Beispiel für die Ausführung des Programms zu sehen, geben Sie Folgendes ein:
example (ClockstaR2)Der Rest dieses Tutorials verwendet den Ordner „clockstar_example_data“.
Der erste Schritt besteht darin, die Genbäume für jede der Ausrichtungen zu erhalten. Dazu verwenden wir eine Baumtopologie und optimieren die Zweiglängen mithilfe jedes einzelnen Gen-Alignments, in diesem Fall A1.fasta bis C3.fasta. Wenn Sie die Genbäume haben, speichern Sie sie im Newick-Format in einer Datei und fahren Sie mit dem nächsten Schritt fort (interaktives Ausführen von Clockstar).
Geben Sie den folgenden Code in die R-Eingabeaufforderung ein und drücken Sie die Eingabetaste:
optim . trees . interactive ()Wenn Sie eine Fehlermeldung zur Installation des Pakets Phangorn erhalten, verwenden Sie bitte diesen Code und wiederholen Sie dann optim.trees.interactive()
install . packcages ( " phangorn " )ClockstaR gibt die folgende Meldung aus:
Please drag a folder with the data subsets and a tree topology . The files should be in FASTA format, and the trees in NEWICKZiehen Sie den Ordner „clockstar_example_data“ auf die R-Konsole und geben Sie die Eingabetaste ein. Beachten Sie, dass der Ordner nur die Alingments im FASTA-Format und die Baumtopologie in NEWICK enthalten sollte. Sie sehen die folgende Meldung:
What should be the name of the file to save the optimised trees ?Geben Sie den Namen der Datei für die optimierten Bäume ein. In diesem Fall verwenden wir „example.trees“
example . treesAn dieser Stelle wird ClockstaR fragen, ob für jedes Gen ein separates Substitutionsmodell oder in allen Fällen JC verwendet werden soll. Da diese Daten unter dem JC simuliert wurden, geben wir „n“ ein und drücken die Eingabetaste. Geben Sie „y“ ein, um jedes Substitutionsmodell separat anzugeben.
Nachdem Sie „n“ eingegeben und die Eingabetaste gedrückt haben, wird ClockstaR gestartet. Die Genbäume werden im Grafikgerät gedruckt. Wenn der angegebene Baum verwurzelt war, werden möglicherweise auch einige Warnungen ausgegeben, die ignoriert werden können.
Öffnen Sie den Ordner „clockstar_example_data“. Sie finden eine Datei mit dem Namen „example.trees“, wie in einigen Schritten oben angegeben. Öffnen Sie example.trees in einem Texteditor. Es enthält jeden Genbaum und die Baumnamen entsprechend den Namen der Genanordnungen. Es sollte ungefähr so aussehen:
A1 . fasta (( t1 : 0.01504695462 ,( t2 : 0.00987 ...
A2 . fasta (( t1 : 0.01520523401 ,( t2 : 0.01317 ...
A3 . fasta (( t1 : 0.01519309467 ,( t2 : 0.01092 ...
.
.
.Diese Datei mit Bäumen wird für den nächsten Schritt verwendet.
Für diesen Schritt ist es notwendig, die Genbäume in einer Datei zu haben, wie sie beispielsweise im vorherigen Schritt erhalten wurde.
Öffnen Sie R und laden Sie ClockstaR wie oben gezeigt. Geben Sie an der Eingabeaufforderung den folgenden Code ein:
clockstar . interactive ()ClockstaR gibt die folgende Meldung aus:
please drag or type in the path to your gene trees file in NEWICK format :Ziehen Sie die Datei mit den Genbäumen auf die R-Konsole. Wenn Sie dem vorherigen Schritt gefolgt sind, heißt die Datei example.trees. Geben Sie Enter ein.
Abhängig von den Paketen, die Sie installiert haben, fragt ClockstaR möglicherweise, ob es parallel laufen soll. Dies ist für große Datenmengen effizient. Für die Beispieldaten wird es jedoch keinen großen Unterschied machen. Geben Sie daher „n“ ein, wenn Sie diese Meldung sehen, und geben Sie dann die Eingabetaste ein:
Packages foreach and doParallel are available for parallel computation
Should we run ClockstaR in parallel (y / n) ? (This is good for large data sets)Clockstar startet nun. Die Ausgabe auf dem Bildschirm sollte etwa so aussehen:
[ 1 ] " Calculating sBSDmin distances between all pairs of trees "
[ 1 ] " Estimating tree distances "
[ 1 ] " estimating distances 1 of 11 "
[ 1 ] " estimating distances 2 of 11 "
[ 1 ] " estimating distances 3 of 11 "
[ 1 ] " estimating distances 4 of 11 "
[ 1 ] " estimating distances 5 of 11 "
.
.
.Nach der Schätzung der Baumabstände (in der Originalveröffentlichung beschrieben) gibt ClockstaR die folgende Meldung aus:
" I finished calculating the sBSDmin distances between trees "
The settings for clustering with ClockstaR are :
PAM clustering algorithm
K from 1 to number of data subsets - 1
SEmax criterion to select the optimal k
500 bootstrap replicates
Are these correct ? (y / n)Dies sind die Einstellungen für den Clustering-Algorithmus. Sie sind für die meisten Datensätze geeignet. In diesem Beispiel können wir also „y“ eingeben und dann eingeben. Durch Eingabe von „n“ können wir diese Einstellungen ändern. Weitere Einzelheiten finden Sie in Kaufman und Rousseeuw (2009).
ClockstaR führt nun den Clustering-Algorithmus aus. Am Ende wird die beste Anzahl an Partitionen ausgedruckt und gefragt, ob die Ergebnisse in einer PDF-Datei gespeichert werden sollen:
[ 1 ] " ClockstaR has finished running "
[ 1 ] " The best number of partitions for your data set is: 3 "
Do you wish to save the results in a pdf file ? (y / n)Geben Sie „y“ ein und geben Sie dann ein.
ClockstaR fragt dann nach dem Namen der Ausgabedateien:
What should be the name and path of the output file ?Geben Sie für dieses Beispiel „example_run“ ein und geben Sie ein, es kann jedoch ein beliebiger Name verwendet werden.
Öffnen Sie nun den Ordner „clockstar_example_data“ und öffnen Sie die beiden PDF-Dateien „example_run_gapstats.pdf“ und „example_run_matrix.pdf“.
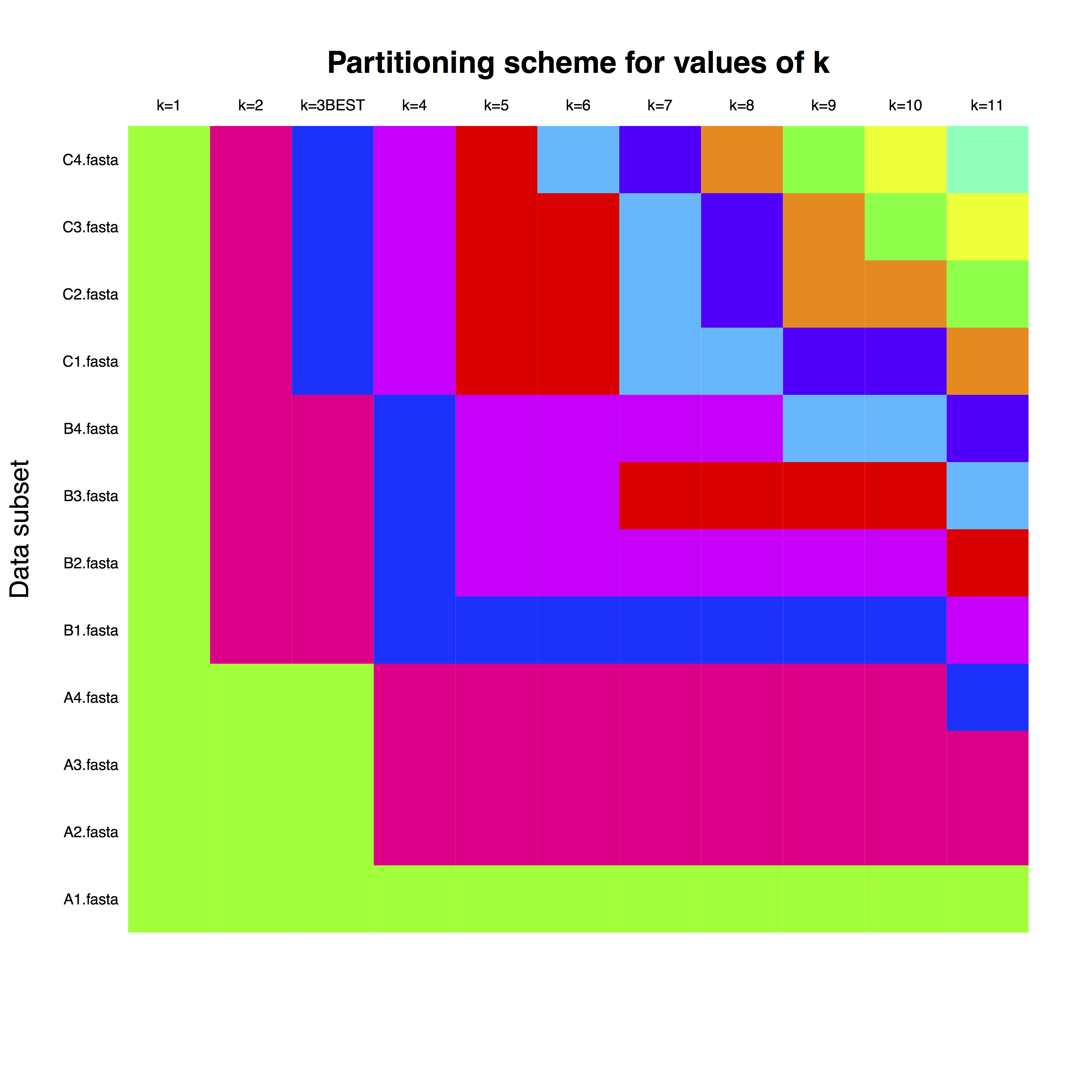
example_run_matrix ist eine Matrix, in der Zeilen jedem Gen entsprechen, wie in den FASTA-Dateien benannt. Die Spalten geben die Anzahl der Partitionen an und die Farben stellen die Zuordnung jedes Gens zur Uhrenpartition dar. Beispielsweise kann man für k = 3, was die beste Anzahl an Partitionen darstellt, separate Uhrenpartitionen für Gene mit den Buchstaben A, B und C verwenden.
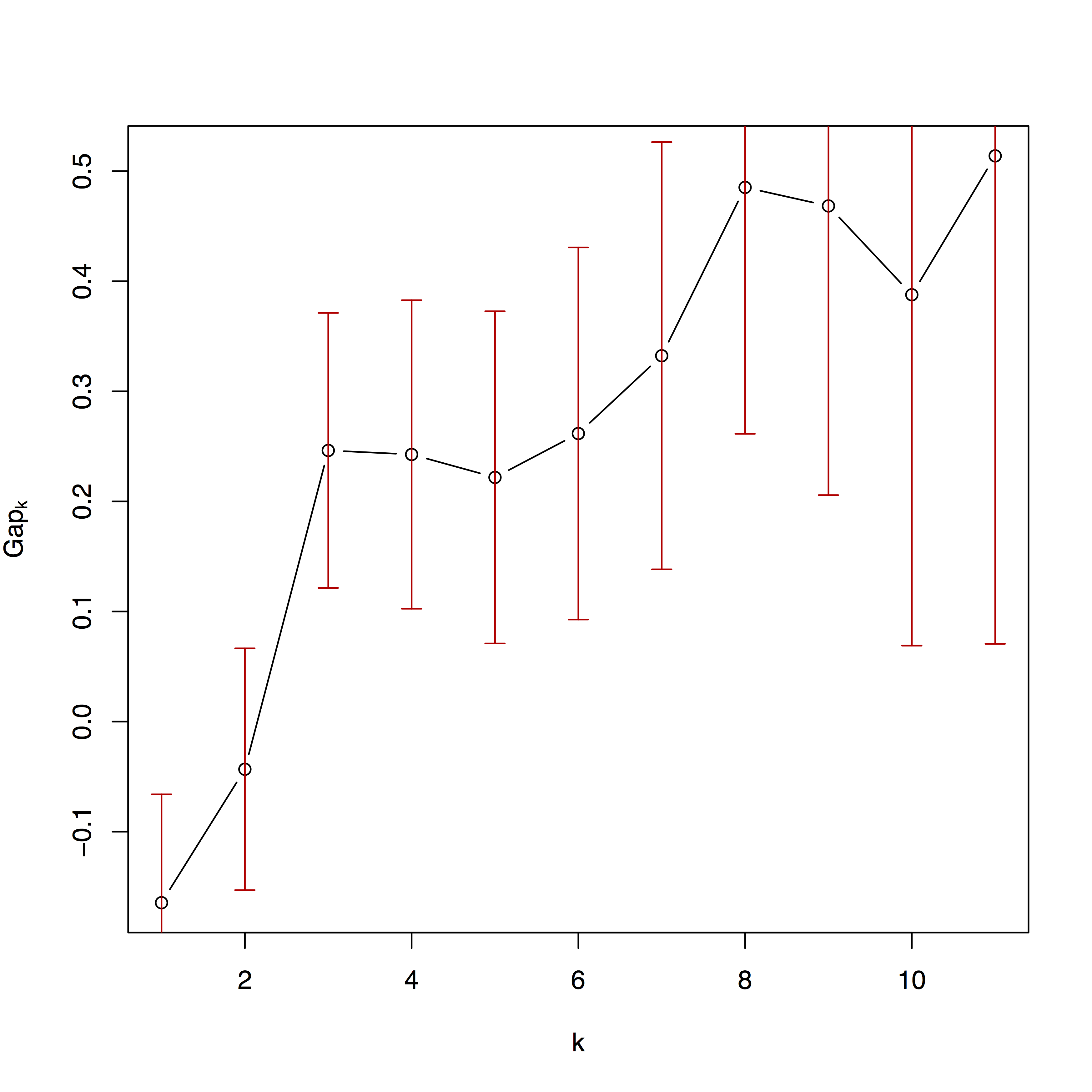
Das zweite Diagramm zeigt die Anpassung der Clustering-Algorithmen über eine unterschiedliche Anzahl von Partitionen hinweg. Weitere Einzelheiten finden Sie in Kaufman und Rousseeuw (2009) und in der Dokumentation zum Paketcluster.
ClockstaR kann mit anderen benutzerdefinierten Einstellungen ausgeführt werden. Weitere Einzelheiten entnehmen Sie bitte der Dokumentation oder schreiben Sie mir bei Fragen an sebastian.duchene[at]sydney.edy.au.
Das Logo wurde von Jun Tong entworfen
Duchene, S. & Ho, SY (2014a). Verwendung mehrerer Relaxed-Clock-Modelle zur Schätzung evolutionärer Zeitskalen aus DNA-Sequenzdaten. Molekulare Phylogenetik und Evolution (77): 65-70.
Duchene, S., Molak, M. & Ho, SY (2014b). ClockstaR: Auswahl der Anzahl entspannter Uhrenmodelle in der molekularen phylogenetischen Analyse. Bioinformatik 30 (7): 1017-1019.
Kaufman, L. & Rousseeuw, PJ (2009). Gruppen in Daten finden: eine Einführung in die Clusteranalyse (Bd. 344). John Wiley & Söhne.


