reference_database_creator
bug fix --in-silico-pcr --untrimmed
KRABBEN ( C reating R Referenzdatenbanken für A plicon- B ased S equencing) ist ein vielseitiges Softwareprogramm, das kuratierte Referenzdatenbanken für die metagenomische Analyse generiert. Der CRABS-Workflow besteht aus sieben Modulen: (i) Herunterladen von Daten aus Online-Repositories; (ii) heruntergeladene Daten in das CRABS-Format importieren; (iii) Amplikonregionen durch In-silico -PCR-Analyse extrahieren; (iv) Amplikons ohne Primer-Bindungsregionen durch Alignments mit in silico extrahierten Barcodes abzurufen; (v) die lokale Datenbank über mehrere Filterparameter kuratieren und unterteilen; (vi) Exportieren der lokalen Datenbank in verschiedene Formate gemäß den Anforderungen des taxonomischen Klassifikators; und (vi) Nachbearbeitungsfunktionen, dh Visualisierungen, um die lokale Referenzdatenbank zu erkunden und einen zusammenfassenden Überblick darüber zu geben. Diese sieben Module sind in achtzehn Funktionen unterteilt und werden im Folgenden beschrieben. Darüber hinaus wird für jede der achtzehn Funktionen Beispielcode bereitgestellt. Abschließend finden Sie am Ende dieses README-Dokuments ein Tutorial zum Erstellen einer lokalen Hai-Referenzdatenbank für den MiFish-E-Primersatz, um ein Beispielskript als Referenz bereitzustellen.
Wir freuen uns, Ihnen mitteilen zu können, dass CRABS auf der Grundlage des Benutzerfeedbacks ein umfassendes Update und eine Code-Neugestaltung erfahren hat, die hoffentlich das Benutzererlebnis beim Aufbau Ihrer ganz eigenen lokalen Referenzdatenbank verbessern wird!
Nachfolgend finden Sie eine Liste der Funktionen und Verbesserungen, die CRABS v 1.0.0 hinzugefügt wurden:
CRABS v 1.0.0 kann jetzt manuell heruntergeladen werden, indem dieses GitHub-Repository geklont wird (ausführliche Informationen finden Sie unter 4.1 Manuelle Installation). Wir werden den Docker-Container und das Conda-Paket so schnell wie möglich aktualisieren, um eine einfache Installation der neuesten Version zu ermöglichen.
Wenn Sie CRABS in Ihren Forschungsprojekten verwenden, zitieren Sie bitte das folgende Papier:
[Jeunen, G.-J., Dowle, E., Edgecombe, J., von Ammon, U., Gemmell, N. J., & Cross, H. (2022). crabs—A software program to generate curated reference databases for metabarcoding sequencing data. Molecular Ecology Resources, 00, 1– 14.](https://doi.org/10.1111/1755-0998.13741)
CRABS ist ein reines Befehlszeilen-Toolkit, das in typischen Unix/Linux-Umgebungen läuft und ausschließlich in Python3 geschrieben ist. Allerdings nutzt CRABS das Subprocess- Modul in Python, um mehrere Befehle in Bash-Syntax auszuführen, um Python-spezifische Besonderheiten zu umgehen und die Ausführungsgeschwindigkeit zu erhöhen. Wir bieten drei Möglichkeiten zur Installation von CRABS. Für die aktuellste Version von CRABS empfehlen wir die manuelle Installation durch Klonen dieses GitHub-Repositorys und die separate Installation von 10 Abhängigkeiten (Installationsanweisungen für alle Abhängigkeiten finden Sie in 4.1 Manuelle Installation). CRABS kann auch über Docker und Conda installiert werden. Beide Methoden ermöglichen eine einfache Installation, indem alle Abhängigkeiten automatisch mitinstalliert werden. Unser Ziel ist es, den Docker-Container und das Conda-Paket auf dem neuesten Stand zu halten, allerdings kann es zu einer gewissen Verzögerung bei der Aktualisierung auf die neueste Version kommen, insbesondere beim Conda-Paket. Nachfolgend finden Sie Einzelheiten zu allen drei Ansätzen.
Klonen Sie für die manuelle Installation zunächst das CRABS-Repository. Für diesen Schritt muss GitHub in der Befehlszeile verfügbar sein (Installationsanweisungen für GitHub).
git clone https://github.com/gjeunen/reference_database_creator.git
Abhängig von Ihren Einstellungen muss CRABS möglicherweise auf Ihrem System ausführbar gemacht werden. Dies kann mit dem folgenden Code erreicht werden.
chmod +x reference_database_creator/crabs
Sobald CRABS installiert ist, müssen wir sicherstellen, dass alle Abhängigkeiten installiert und global zugänglich sind. Die neueste Version von CRABS (Version v 1.0.0 ) läuft auf Python 3.11.7 (oder einer beliebigen kompatiblen Version zu 3.11.7) und basiert auf fünf Python-Modulen, die möglicherweise nicht standardmäßig in Python enthalten sind, sowie fünf externen Softwareprogrammen. Alle Abhängigkeiten sind unten aufgeführt, zusammen mit einem Link zur Installationsanleitung. Die für jedes Modul und Softwareprogramm angegebenen Versionsnummern entsprechen denen, auf denen CRABS entwickelt wurde. Es könnten jedoch auch kompatible Versionen von jedem verwendet werden.
Python-Module:
Externe Softwareprogramme:
Sobald CRABS und alle Abhängigkeiten installiert sind, kann CRABS mithilfe des folgenden Codes im gesamten Betriebssystem zugänglich gemacht werden.
export PATH="/path/to/crabs/folder:$PATH"
Ersetzen Sie /path/to/crabs/folder durch den tatsächlichen Pfad zum GitHub-Repository-Ordner auf dem Betriebssystem, d. h. den Ordner, der während des obigen Befehls git clone erstellt wurde. Durch das Hinzufügen des export zur .bash_profile- oder .bashrc-Datei wird CRABS jederzeit global zugänglich gemacht.
Docker ist ein Open-Source-Projekt, das die Bereitstellung von Softwareanwendungen in „Containern“ ermöglicht, die von Ihrem Computer isoliert sind und über ein virtuelles Host-Betriebssystem namens Docker Engine ausgeführt werden. Der Hauptvorteil der Ausführung von Docker gegenüber virtuellen Maschinen besteht darin, dass sie viel weniger Ressourcen verbrauchen. Diese Isolation bedeutet, dass Sie einen Docker-Container auf den meisten Betriebssystemen ausführen können, einschließlich Mac, Windows und Linux. Möglicherweise müssen Sie ein kostenloses Konto einrichten, um Docker Desktop nutzen zu können. Dieser Link bietet eine schöne Einführung in die Grundlagen der Verwendung von Docker. Hier ist ein Link, der Ihnen den Einstieg und die Orientierung im Docker-Multiversum erleichtert.
Es sind nur zwei Schritte erforderlich, um die Crabs auf Ihrem Computer zum Laufen zu bringen. Installieren Sie zunächst Docker Desktop auf Ihrem Computer, was für die meisten Benutzer kostenlos ist. Hier sind die Anweisungen für Mac ; Hier sind die Anweisungen für Windows-Computer und hier sind die Anweisungen für Linux (die meisten wichtigen Linux-Plattformen werden unterstützt). Sobald Sie Docker Desktop installiert und ausgeführt haben (die Desktop-Anwendung muss ausgeführt werden, damit Sie Docker-Befehle in der Befehlszeile verwenden können), müssen Sie nur noch unser Crabs-Image „ziehen“ und schon kann es losgehen:
docker pull quay.io/swordfish/crabs:0.1.7
Während die Installation einer Docker-Anwendung einfach ist, kann die Verwendung dieser Anwendungen zunächst etwas schwierig sein. Um Ihnen den Einstieg zu erleichtern, haben wir einige Beispielbefehle mit der Docker-Version von crabs bereitgestellt. Diese Beispiele finden Sie im Ordner docker_intro in diesem Repo . Anhand dieser Beispiele sollten Sie in der Lage sein, die Einrichtung einer gesamten Referenzdatenbank durchzuführen und bereit zu sein. Wir werden diese Beispiele weiter ausbauen und dies in vielen verschiedenen Situationen testen. Bitte stellen Sie Fragen und geben Sie Feedback auf der Registerkarte „Probleme“.
Um das Conda-Paket zu installieren, müssen Sie zuerst Conda installieren. Einzelheiten finden Sie unter diesem Link. Wenn Conda bereits installiert ist, empfiehlt es sich, das Conda-Tool vor der Installation von CRABS mit conda update conda zu aktualisieren.
Führen Sie nach der Installation von Conda die folgenden Schritte aus, um CRABS und alle Abhängigkeiten zu installieren. Achten Sie darauf, die Befehle in der unten aufgeführten Reihenfolge einzugeben.
conda create -n CRABS
conda activate CRABS
conda config --add channels bioconda
conda config --add channels conda-forge
conda install -c bioconda crabs
Sobald Sie den Installationsbefehl eingegeben haben, verarbeitet Conda die Anfrage (dies kann etwa eine Minute dauern), zeigt dann alle Pakete und Programme an, die installiert werden, und fordert Sie zur Bestätigung auf. Geben Sie y um die Installation zu starten. Danach sollte CRABS einsatzbereit sein.
Wir haben diese Installation auf Mac- und Linux-Systemen getestet. Wir haben das Windows-Subsystem für Linux (WSL) noch nicht getestet.
Verwenden Sie den folgenden Code, um zu überprüfen, ob CRABS erfolgreich installiert wurde, und rufen Sie die Hilfeinformationen auf.
crabs -hDie Hilfeinformationen unterteilen die achtzehn Funktionen in verschiedene Gruppen, wobei jede Gruppe oben die Funktion und darunter die erforderlichen und optionalen Parameter auflistet.

CRABS enthält sieben Module, die achtzehn Funktionen umfassen:
Modul 1: Daten aus Online-Repositories herunterladen
--download-taxonomy : NCBI-Taxonomieinformationen herunterladen;--download-bold : Sequenzdaten aus der Barcode of Life-Datenbank (BOLD) herunterladen;--download-embl : Sequenzdaten vom Europäischen Nukleotidarchiv (ENA; EMBL) herunterladen;--download-mitofish : Sequenzdaten aus der MitoFish-Datenbank herunterladen;--download-ncbi : Sequenzdaten vom National Center for Biotechnology Information (NCBI) herunterladen.Modul 2: Heruntergeladene Daten in das CRABS-Format importieren
--import : Heruntergeladene Sequenzen oder benutzerdefinierte Barcodes in das CRABS-Format importieren;--merge : Verschiedene CRABS-formatierte Dateien in einer einzigen Datei zusammenführen.Modul 3: Extrahieren von Amplikonregionen durch In-silico -PCR-Analyse
--in-silico-pcr : Extrahieren Sie Amplifikate aus heruntergeladenen Daten, indem Sie Primer-Bindungsbereiche lokalisieren und entfernen.Modul 4: Amplikons ohne Primer-Bindungsregionen abrufen
--pairwise-global-alignment : Rufen Sie Amplikons ohne Primer-Bindungsregionen ab, indem Sie heruntergeladene Sequenzen an in silico extrahierten Barcodes ausrichten.Modul 5: Kuratieren und Unterteilen der lokalen Datenbank über mehrere Filterparameter
--dereplicate : doppelte Sequenzen verwerfen;--filter : Sequenzen über mehrere Filterparameter verwerfen;--subset : Teilmenge der lokalen Datenbank, um bestimmte taxonomische Gruppen beizubehalten oder auszuschließen.Modul 6: Exportieren Sie die lokale Datenbank
--export : Exportiert die CRABS-formatierte Datenbank in verschiedene Formate entsprechend den Anforderungen des zu verwendenden taxonomischen Klassifikators.Modul 7: Nachbearbeitungsfunktionen zur Erkundung und Bereitstellung eines zusammenfassenden Überblicks über die lokale Referenzdatenbank
--diversity-figure : erstellt ein horizontales Balkendiagramm, das die Anzahl der Arten und Sequenzgruppen pro angegebener Ebene anzeigt, die in der Referenzdatenbank enthalten sind;--amplicon-length-figure : erstellt ein Liniendiagramm, das die Längenverteilungen der Amplikons getrennt nach taxonomischen Gruppen darstellt;--phylogenetic-tree : erstellt einen phylogenetischen Baum mit Barcodes aus der Referenzdatenbank für eine Zielliste von Arten;--amplification-efficiency-figure : erstellt ein Balkendiagramm, das Fehlpaarungen in den Primer-Bindungsregionen anzeigt;--completeness-table : Erstellt eine Tabelle mit der Barcode-Verfügbarkeit für taxonomische Gruppen.Erste Sequenzierungsdaten können von CRABS aus vier Online-Repositories heruntergeladen werden, darunter (i) BOLD, (ii) EMBL, (iii) MitoFish und NCBI. Ab Version v 1.0.0 ist das Herunterladen von Daten aus jedem Repository in eine eigene Funktion aufgeteilt. Darüber hinaus formatiert CRABS die Daten nach dem Herunterladen nicht automatisch, um die Flexibilität zu erhöhen und das Debuggen zu ermöglichen, wenn der Download der Daten fehlschlägt.
CRABS kann nicht nur Sequenzdaten herunterladen, sondern auch die NCBI-Taxonomieinformationen herunterladen, die CRABS zum Erstellen der taxonomischen Abstammungslinie für jede Sequenz verwendet.
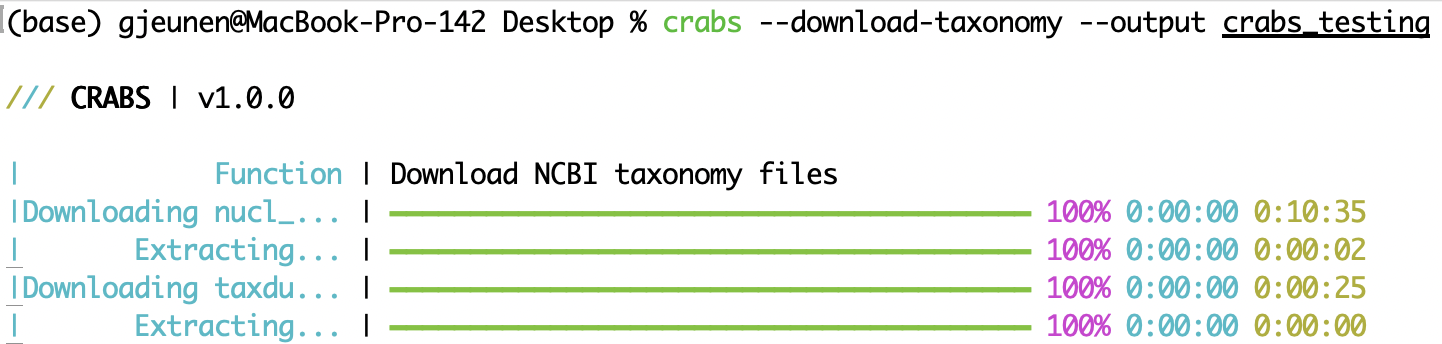
--download-taxonomy Um jeder heruntergeladenen Sequenz in der Referenzdatenbank (siehe 5.2 Modul 2) eine taxonomische Abstammungslinie zuzuordnen, müssen die taxonomischen Informationen heruntergeladen werden. CRABS nutzt die Taxonomie von NCBI und lädt drei spezifische Dateien auf Ihren Computer herunter: (i) eine Datei, die Zugangsnummern mit taxonomischen IDs verknüpft ( nucl_gb.accession2taxid ), (ii) eine Datei mit Informationen über den phylogenetischen Namen, der jeder taxonomischen ID zugeordnet ist ( names.dmp ) und (iii) eine Datei mit Informationen darüber, wie taxonomische IDs verknüpft sind ( nodes.dmp ). Das Ausgabeverzeichnis für die heruntergeladenen Dateien kann mit dem Parameter --output angegeben werden. Um entweder die Datei „nucl_gb.accession2taxid“ oder die Dateien „names.dmp“ und „nodes.dmp“ auszuschließen, kann der Parameter --exclude acc2tax bzw. --exclude taxdump angegeben werden. Der erste Code unten lädt keine Datei herunter, da sowohl acc2tax als auch taxdump für den Parameter --exclude bereitgestellt werden. Die zweite Codezeile lädt alle drei Dateien in das Unterverzeichnis --output crabs_testing herunter. Der Screenshot unten zeigt, was bei der Ausführung dieser Codezeile auf der Konsole ausgegeben wird.
crabs --download-taxonomy --exclude 'acc2taxid,taxdump'
crabs --download-taxonomy --output crabs_testing
--download-bold BOLD-Sequenzen werden über die BOLD-Website heruntergeladen. Die Ausgabedatei, die als zweizeiliges Fasta-Dokument aufgebaut ist, kann mit dem Parameter --output angegeben werden. Benutzer können mithilfe des Parameters --taxon angeben, welche taxonomische Gruppe heruntergeladen werden soll. Wir empfehlen, eine einfache for-Schleife zu schreiben (Beispiel siehe unten), wenn Benutzer mehrere taxonomische Gruppen herunterladen möchten, um so die Menge der von BOLD pro Instanz herunterzuladenden Daten zu begrenzen. Wenn jedoch nur eine begrenzte Anzahl taxonomischer Gruppen von Interesse ist, können taxonomische Gruppennamen auch durch | getrennt werden (Beispiel siehe unten). Wir empfehlen Benutzern außerdem zu prüfen, ob der herunterzuladende taxonomische Gruppenname im BOLD-Archiv aufgeführt ist oder ob alternative Namen verwendet werden müssen. Wenn Sie beispielsweise --taxon Chondrichthyes angeben, werden nicht alle Knorpelfischsequenzen von BOLD heruntergeladen, da dieser Klassenname nicht in BOLD aufgeführt ist. Benutzer sollten in diesem Fall lieber --taxon Elasmobranchii verwenden. Benutzer können den Download auch auf einen bestimmten genetischen Marker beschränken, indem sie den Parameter --marker angeben. Wenn mehrere genetische Marker von Interesse sind, sollten die Markernamen durch | getrennt werden . Die vier wichtigsten DNA-Barcode-Marker auf BOLD sind COI-5P , ITS , matK und rbcL . Bei der Eingabe für den Parameter --marker wird die Groß-/Kleinschreibung beachtet.
Empfohlener Ansatz: Eine einfache for-Schleife zum Herunterladen von Daten aus BOLD für mehrere taxonomische Gruppen (empfohlener Ansatz). Der folgende Code lädt zunächst Daten für Elasmobranchii herunter, gefolgt von Sequenzen, die Mammalia zugeordnet sind. Heruntergeladene Daten werden in das Unterverzeichnis --output crabs_testing geschrieben und in zwei separaten Dateien abgelegt, die angeben, welche Daten zu welcher taxonomischen Gruppe gehören, dh crabs_testing/bold_Elasmobranchii.fasta und crabs_testing/bold_Mammalia.fasta .
for taxon in Elasmobranchii Mammalia; do crabs --download-bold --taxon ${taxon} --output crabs_testing/bold_${taxon}.fasta; done

Alternative Möglichkeit: Neben der empfohlenen for-Schleife können mehrere Taxonnamen gleichzeitig bereitgestellt werden, indem die Namen durch | getrennt werden .
crabs --download-bold --taxon 'Elasmobranchii|Mammalia' --output crabs_testing/bold_elasmobranchii_mammalia.fasta
--download-embl Sequenzen von EMBL werden über die ENA-FTP-Site heruntergeladen. EMBL-Dateien werden zunächst im Format „.fasta.gz“ heruntergeladen und nach Abschluss des Downloads automatisch entpackt. Diese Datenbank bietet im Vergleich zu BOLD oder NCBI nicht so viel Flexibilität hinsichtlich des selektiven Herunterladens. Vielmehr sind die EMBL-Daten in 15 Steuerabteilungen gegliedert, die separat heruntergeladen werden können. Die herunterzuladende Steuerabteilung kann mit dem Parameter --taxon angegeben werden. Da jede Steuerabteilung in mehrere Dateien aufgeteilt ist, wird nach dem Namen ein * angegeben, um alle Dateien herunterzuladen. Benutzer können eine bestimmte Datei auch herunterladen, indem sie den vollständigen Dateinamen eingeben. Nachfolgend finden Sie eine Liste aller 15 Steueraufteilungsoptionen. Das Ausgabeverzeichnis und der Dateiname können mit dem Parameter --output angegeben werden.
Liste der Steuerabteilungen:
crabs --download-embl --taxon 'mam*' --output crabs_testing/embl_mam.fasta
--download-mitofish CRABS kann auch die MitoFish-Datenbank herunterladen. Bei dieser Datenbank handelt es sich um eine einzelne zweizeilige Fasta-Datei. Das Ausgabeverzeichnis und der Dateiname können mit dem Parameter --output angegeben werden.
crabs --download-mitofish --output crabs_testing/mitofish.fasta

--download-ncbi Sequenzen aus der NCBI-Datenbank werden über die Entrez Programming Utilities heruntergeladen. NCBI ermöglicht das Herunterladen von Daten aus verschiedenen Datenbanken, die Benutzer mit dem Parameter --database angeben können. Für die meisten Benutzer ist die --database nucleotide am besten zum Aufbau einer lokalen Referenzdatenbank geeignet.
Um die von NCBI herunterzuladenden Daten anzugeben, stellen Benutzer eine Suche über den Parameter --query bereit. Es kann schwierig sein, gute NCBI-Suchen zu erstellen. Eine gute Möglichkeit, eine Suchabfrage zu erstellen, ist die Verwendung des NCBI-Webseiten-Suchfensters. Führen Sie über diesen Link zunächst eine erste Suche durch und drücken Sie die Eingabetaste. Dadurch gelangen Sie zur Ergebnisseite, auf der Sie Ihre Suche weiter verfeinern können. Im Screenshot unten haben wir die Suche weiter verfeinert, indem wir die Sequenzlänge auf 100–25.000 bp begrenzt haben und nur mitochondriale Sequenzen einbezogen haben. Benutzer können den Text in das Feld „Suchdetails“ auf der Website kopieren und einfügen und ihn in Anführungszeichen für den Parameter --query angeben. Ein weiterer Vorteil der Verwendung des NCBI-Webseiten-Suchfensters besteht darin, dass auf der Webseite angezeigt wird, wie viele Sequenzen mit Ihrer Suchanfrage übereinstimmen, was mit der Anzahl der von CRABS gemeldeten Sequenzen übereinstimmen sollte. Diese Webseite bietet ein weiteres kurzes Tutorial zur Verwendung der Suchfunktion auf der NCBI-Webseite, das unser Team für zusätzliche Informationen geschrieben hat.
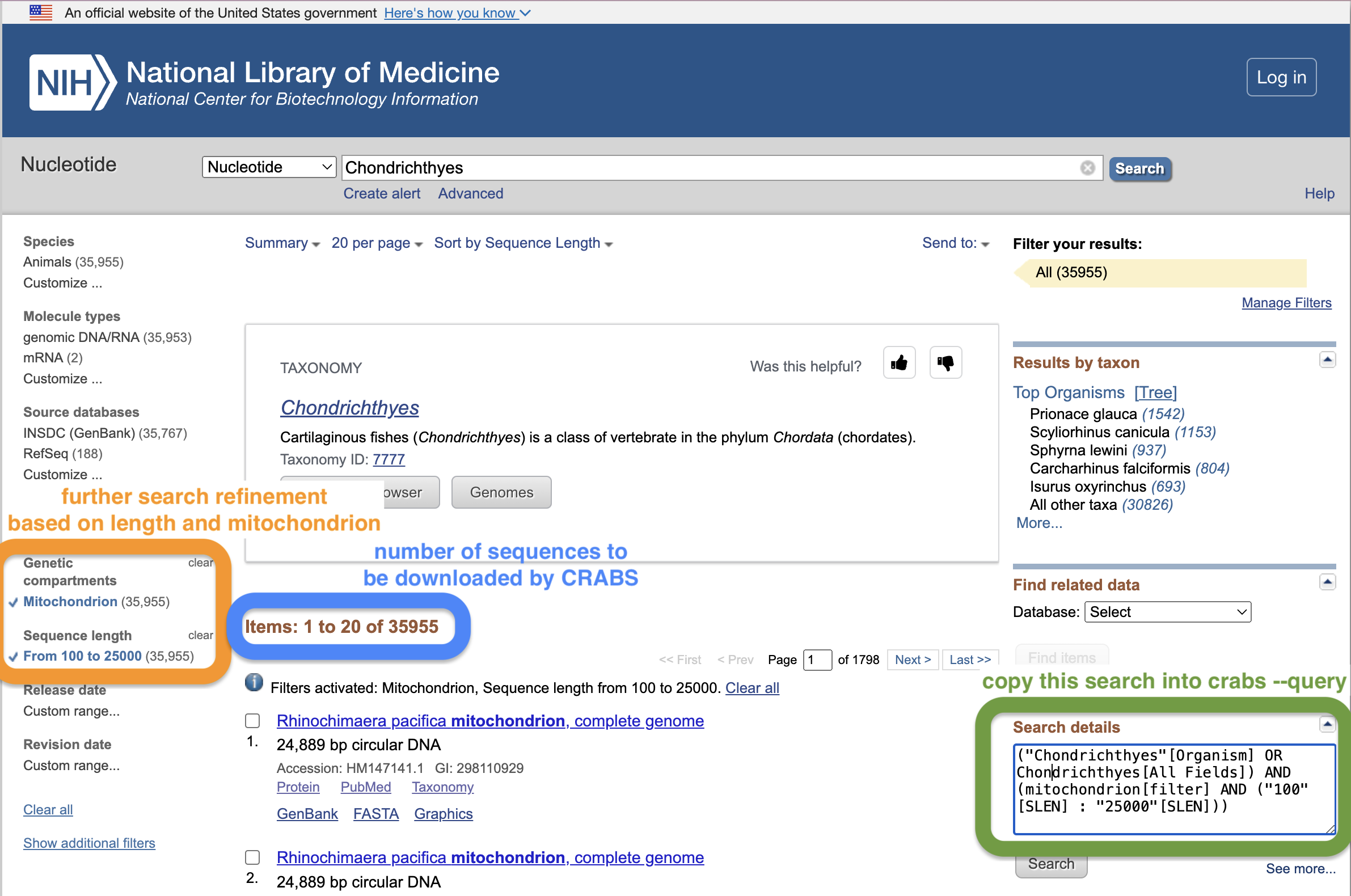
Neben der Suchabfrage ( --query ) können Benutzer den Suchbegriff weiter einschränken, indem sie mithilfe des Parameters --species Sequenzdaten für eine Liste von Arten herunterladen. Der Parameter --species akzeptiert entweder eine Eingabezeichenfolge mit durch + getrennten Artennamen oder eine TXT-Eingabedatei mit einem einzelnen Artennamen pro Zeile im Dokument. Der Parameter --batchsize bietet Benutzern die Möglichkeit, Sequenzen in N-Batches von der NCBI-Website herunterzuladen. Dieser Parameter ist standardmäßig auf 5.000 eingestellt. Es wird nicht empfohlen, diesen Wert über 5.000 zu erhöhen, da die NCBI-Server den Download höchstwahrscheinlich unterbrechen, wenn zu viele Sequenzen gleichzeitig heruntergeladen werden. Mit dem Parameter --email können Benutzer ihre E-Mail-Adresse angeben, die für den Zugriff auf die NCBI-Server erforderlich ist. Abschließend können das Ausgabeverzeichnis und der Dateiname mit dem Parameter --output angegeben werden.
crabs --download-ncbi --query '("Chondrichthyes"[Organism] OR Chondrichthyes[All Fields]) AND (mitochondrion[filter] AND ("100"[SLEN] : "25000"[SLEN]))' --output crabs_testing/ncbi_chondrichthyes.fasta --email [email protected] --database nucleotide

--import Sobald die Daten aus Online-Repositories heruntergeladen wurden, müssen die Dateien mit der Funktion --import in CRABS importiert werden. Das CRABS-Format besteht aus einer einzelnen tabulatorgetrennten Zeile pro Sequenz, die alle Informationen enthält, einschließlich (i) Sequenz-ID, (ii) taxonomischer Name, der aus dem ersten Download analysiert wurde, (iii) NCBI-Taxon-ID-Nummer, (iv) taxonomische Abstammung gemäß NCBI-Taxonomie , und (v) die Sequenz. CRABS wird versuchen, die NCBI-Zugangsnummer für jede Sequenz als Sequenz-ID zu erhalten. Wenn die Sequenz keine Zugangsnummer enthält, also nicht bei NCBI hinterlegt ist, generiert CRABS eindeutige Sequenz-IDs im folgenden Format: crabs_*[num]*_taxonomic_name . Das Format des Eingabedokuments wird mit dem Parameter --import-format angegeben und gibt den Namen des Repositorys an, aus dem die Daten heruntergeladen wurden, z. B. BOLD , EMBL , MITOFISH oder NCBI . Die von CRABS erstellte taxonomische Abstammungslinie basiert auf der NCBI-Taxonomie und CRABS benötigt die drei Dateien, die mit der Funktion --download-taxonomy heruntergeladen wurden, also --names , --nodes und --acc2tax . Ab Version v 1.0.0 ist CRABS in der Lage, Synonyme und nicht akzeptierte Namen aufzulösen, um eine größere Anzahl von Sequenzen und Diversität in die lokale Referenzdatenbank aufzunehmen. Die taxonomischen Ränge, die in die taxonomische Abstammungslinie einbezogen werden sollen, können mit den Parametern --ranks angegeben werden. Obwohl jeder taxonomische Rang einbezogen werden kann, empfehlen wir die Verwendung der folgenden Eingabe, um alle notwendigen Informationen für die meisten taxonomischen Klassifikatoren einzuschließen --ranks 'superkingdom;phylum;class;order;family;genus;species' . Die Ausgabedatei kann mit dem Parameter --output angegeben werden und ist eine einfache TXT-Datei. Im Terminalfenster druckt CRABS die Ergebnisse der Anzahl der importierten Sequenzen sowie aller Sequenzen, für die keine taxonomische Abstammungslinie generiert werden konnte.
crabs --import --import-format bold --input crabs_testing/bold_Elasmobranchii.fasta --names crabs_testing/names.dmp --nodes crabs_testing/nodes.dmp --acc2tax crabs_testing/nucl_gb.accession2taxid --output crabs_testing/crabs_bold.txt --ranks 'superkingdom;phylum;class;order;family;genus;species'

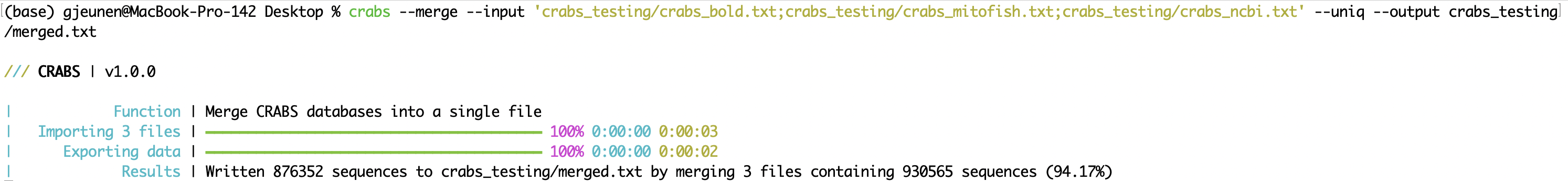
--merge Wenn Sequenzdaten aus mehreren Online-Repositorys heruntergeladen werden, können Dateien nach dem Import (siehe 5.2.1 --import ) mithilfe der Funktion --merge zu einer einzigen Datei zusammengeführt werden. Die zusammenzuführenden Eingabedateien können mit dem Parameter --input eingegeben werden, wobei die Dateien durch ; getrennt werden ; . Es ist möglich, dass eine Sequenz beim Ablegen in verschiedenen Online-Repositories mehrmals heruntergeladen wurde. Durch die Verwendung des Parameters --uniq bleibt nur eine einzige Version jeder Zugangsnummer erhalten. Die Ausgabedatei kann mit dem Parameter --output angegeben werden. Im Terminalfenster druckt CRABS die Ergebnisse der Anzahl der zusammengeführten Sequenzen sowie die Anzahl der beibehaltenen Sequenzen bei Verwendung des Parameters --uniq .
crabs --merge --input 'crabs_testing/crabs_bold.txt;crabs_testing/crabs_mitofish.txt;crabs_testing/crabs_ncbi.txt' --uniq --output crabs_testing/merged.txt
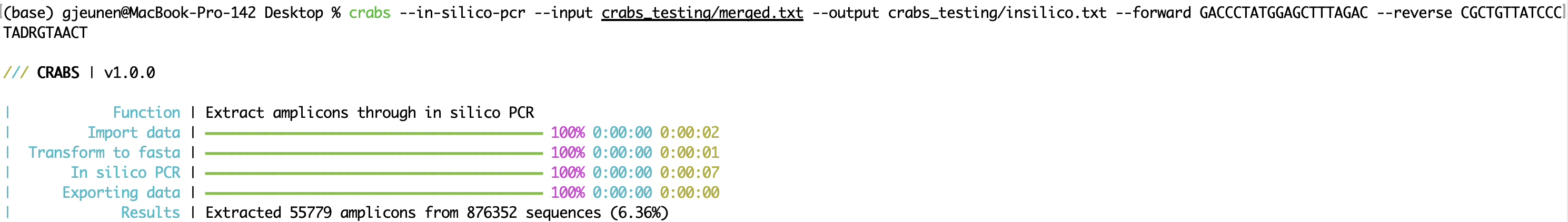
CRABS extrahiert die Amplikonregion des Primersatzes durch Durchführung einer In-silico -PCR (Funktion: --in-silico-pcr ). CRABS verwendet Cutadapt v 4.4 für die In-Silico -PCR, um die Ausführungsgeschwindigkeit von herkömmlichem Python-Code zu erhöhen. Eingabe- und Ausgabedateinamen können mit den Parametern „ --input “ bzw. „ --output “ angegeben werden. Sowohl der Vorwärts- als auch der Rückwärtsprimer sollten in 5'-3'-Richtung mit den Parametern „ --forward “ bzw. „ --reverse “ bereitgestellt werden. CRABS komplementiert den Reverse-Primer rückwärts. Ab Version v 1.0.0 ist CRABS in der Lage, Barcodes in beide Richtungen mithilfe einer einzigen In-silico -PCR-Analyse aufzubewahren. Daher wird kein Rückkomplementierungsschritt und keine erneute Ausführung der In-silico -PCR durchgeführt, wodurch die Ausführungsgeschwindigkeit deutlich erhöht wird. Um Sequenzen beizubehalten, für die keine Primer-Bindungsregionen gefunden werden konnten, kann für den Parameter --untrimmed eine Ausgabedatei angegeben werden. Die maximal zulässige Anzahl von Fehlpaarungen, die in den Primer-Bindungsregionen gefunden werden, kann mit dem Parameter --mismatch angegeben werden, mit einer Standardeinstellung von 4. Schließlich kann die In-silico -PCR-Analyse in CRABS multithreaded ausgeführt werden. Standardmäßig wird die maximale Anzahl an Threads verwendet, Benutzer können jedoch die Anzahl der zu verwendenden Threads mit dem Parameter --threads angeben.
crabs --in-silico-pcr --input crabs_testing/merged.txt --output crabs_testing/insilico.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT
Es ist gängige Praxis, Primer-bindende Regionen aus Referenzsequenzen zu entfernen, wenn sie in einer Online-Datenbank hinterlegt werden. Wenn daher die Referenzsequenz mit demselben Vorwärts- und/oder Rückwärtsprimer generiert wurde, nach dem in der Funktion --in-silico-pcr gesucht wurde, konnte die Funktion --in-silico-pcr die Amplikonregion nicht wiederherstellen Referenzsequenz. Um dieser Möglichkeit Rechnung zu tragen, verfügt CRABS über die Option, ein mit VSEARCH v 2.16.0 implementiertes Pairwise Global Alignment durchzuführen, um Amplikonregionen zu extrahieren, für die die Referenzsequenz nicht die vollständigen Vorwärts- und Rückwärts-Primerbindungsregionen enthält. Um dies zu erreichen, übernimmt die Funktion --pairwise-global-alignment die ursprünglich heruntergeladene Datenbankdatei mithilfe des Parameters --input . Die zu durchsuchende Datenbank ist die Ausgabedatei von --in-silico-pcr und kann mit dem Parameter --amplicons angegeben werden. Die Ausgabedatei kann mit dem Parameter --output angegeben werden. Die Primersequenzen, die nur zur Berechnung der Basenpaarlänge verwendet werden, können mit den Parametern --forward und --reverse festgelegt werden. Da die Ausführung der Funktion --pairwise-global-alignment bei großen Datenbanken lange dauern kann, kann die Sequenzlänge begrenzt werden, um den Prozess mithilfe des Parameters --size-select zu beschleunigen. Der Mindestprozentsatz für Identität und Abfrageabdeckung kann mit den Parametern --percent-identity bzw. --coverage angegeben werden. --percent-identity sollte als Prozentwert zwischen 0 und 1 (z. B. 95 % = 0,95) angegeben werden, während --coverage als Prozentwert zwischen 0 und 100 (z. B. 95 % = 95) angegeben werden sollte. Standardmäßig ist die Funktion --pairwise-global-alignment darauf beschränkt, Sequenzen beizubehalten, bei denen Primersequenzen nicht vollständig in der Referenzsequenz vorhanden sind (Alignment beginnt oder endet innerhalb der Länge des Vorwärts- oder Rückwärtsprimers). Wenn der Parameter --all-start-positions angegeben wird, werden positive Treffer berücksichtigt, wenn das Alignment außerhalb des Bereichs der Primer-Bindungsregionen gefunden wird (von der Funktion --in-silico-pcr aufgrund zu vieler Nichtübereinstimmungen in der Region übersehen). Primer-Bindungsregion). Wir empfehlen die Verwendung von --all-start-positions nicht, da es sehr unwahrscheinlich ist, dass ein Barcode mit dem angegebenen Primersatz der Funktion --in-silico-pcr verstärkt wird, wenn mehr als 4 Fehlpaarungen im Primer vorhanden sind. Bindungsregionen.
crabs --pairwise-global-alignment --input crabs_testing/merged.txt --amplicons crabs_testing/insilico.txt --output crabs_testing/aligned.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT --size-select 10000 --percent-identity 0.95 --coverage 0.95
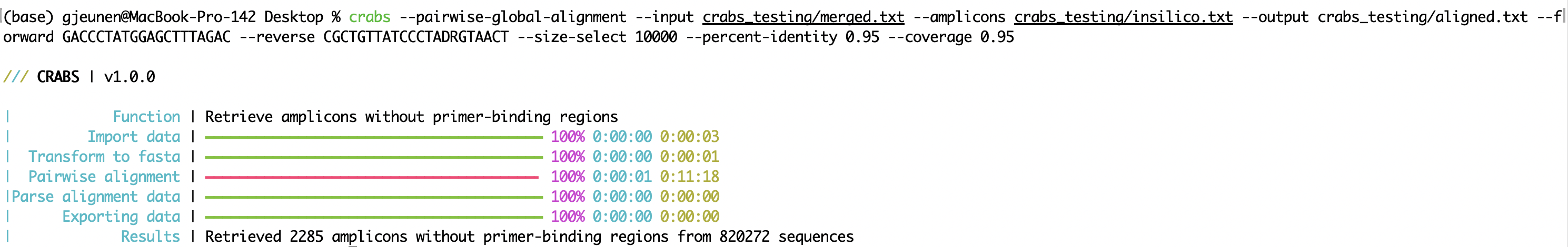
--pairwise-global-alignment Die Ausführung der Funktion --pairwise-global-alignment kann viel Zeit in Anspruch nehmen, wenn CRABS große Sequenzdateien verarbeitet, obwohl Multithreading unterstützt wird. Seit dem Update auf CRABS v 1.0.0 ist von --import bis --export eine identische Dateistruktur vorhanden, wodurch Funktionen in beliebiger Reihenfolge ausgeführt werden können. Wir empfehlen zwar weiterhin, die Reihenfolge des CRABS-Workflows einzuhalten, die Funktion --pairwise-global-alignment kann jedoch erheblich beschleunigt werden, wenn die Funktionen --dereplicate und --filter vor der Funktion --in-silico-pcr ausgeführt werden. Durch die Ausführung dieser Kurationsschritte vor --in-silico-pcr wird die Anzahl der Sequenzen, die von CRABS für die Funktion --pairwise-global-alignment verarbeitet werden müssen, erheblich reduziert.
HINWEIS 1 : Wenn Sie die Funktion --filter vor --in-silico-pcr ausführen, achten Sie bitte darauf, alle Parameter wegzulassen, die sich direkt auf die Sequenz auswirken, da --filter dies auf der gesamten Sequenz und nicht auf dem extrahierten Amplikon basiert . Lassen Sie daher die folgenden Parameter weg: --minimum-length , --maximum-length , --maximum-n .
HINWEIS 2 : Wenn Sie die Funktionen --dereplicate und --filter vor --in-silico-pcr ausführen, wäre es ratsam, beide Funktionen nach --pairwise-global-alignment erneut auszuführen, da die Datenbank jetzt weiter kuratiert werden könnte dass die Amplifikate extrahiert werden.
Sobald alle potenziellen Barcodes für den Primersatz durch die Funktionen --in-silico-pcr und --pairwise-global-alignment extrahiert wurden, kann die lokale Referenzdatenbank innerhalb von CRABS mithilfe verschiedener Funktionen, einschließlich --dereplicate , einer weiteren Kuratierung und Unterteilung unterzogen werden , --filter und --subset .
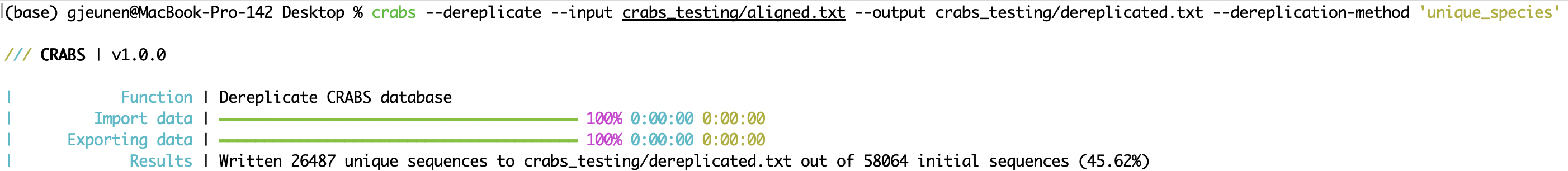
--dereplicate Die erste Kurationsmethode besteht darin, die lokale Referenzdatenbank mithilfe der Funktion --dereplicate zu dereplizieren. Es ist möglich, dass für bestimmte Taxa zu diesem Zeitpunkt mehrere identische Barcodes in der lokalen Referenzdatenbank enthalten sind. Dies kann auftreten, wenn verschiedene Forschungsgruppen identische Sequenzen hinterlegt haben oder wenn die intraspezifische Variation zwischen Sequenzen für ein Taxon nicht im extrahierten Barcode enthalten ist. Es ist am besten, diese identischen Referenzbarcodes zu entfernen, um die Taxonomiezuweisung zu beschleunigen und die Ergebnisse der Taxonomiezuweisung zu verbessern (insbesondere für taxonomische Klassifikatoren, die eine begrenzte Anzahl von Ergebnissen liefern, z. B. BLAST).
Die Eingabe- und Ausgabedateien können mit den Parametern --input bzw. --output angegeben werden. CRABS bietet drei Dereplikationsmethoden, die mit dem Parameter --dereplication-method angegeben werden können, darunter:
crabs --dereplicate --input crabs_testing/aligned.txt --output crabs_testing/dereplicated.txt --dereplication-method 'unique_species'
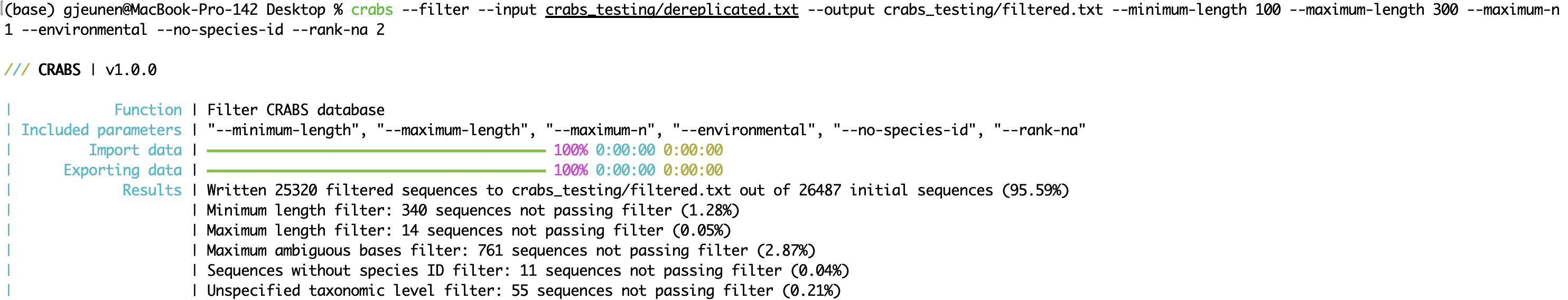
--filter Die zweite Kurationsmethode besteht darin, die lokale Referenzdatenbank mithilfe verschiedener Parameter mithilfe der Funktion --filter zu filtern. Die Eingabe- und Ausgabedateien können mit den Parametern --input bzw. --output angegeben werden. Ab Version v 1.0.0 . CRABS beinhaltet die Filterung basierend auf sechs Parametern, darunter:
--minimum-length : Mindestsequenzlänge, damit ein Amplifikat in der Datenbank beibehalten wird;--maximum-length : maximale Sequenzlänge für ein Amplifikat, das in der Datenbank beibehalten werden soll;--maximum-n : Amplikons mit N oder mehr mehrdeutigen Basen ( N ) verwerfen;--environmental : Umgebungssequenzen aus der Datenbank verwerfen;--no-species-id : Sequenzen verwerfen, für die kein Artname verfügbar ist;--rank-na : Sequenzen mit N oder mehr nicht spezifizierten taxonomischen Ebenen verwerfen. crabs --filter --input crabs_testing/dereplicated.txt --output crabs_testing/filtered.txt --minimum-length 100 --maximum-length 300 --maximum-n 1 --environmental --no-species-id --rank-na 2
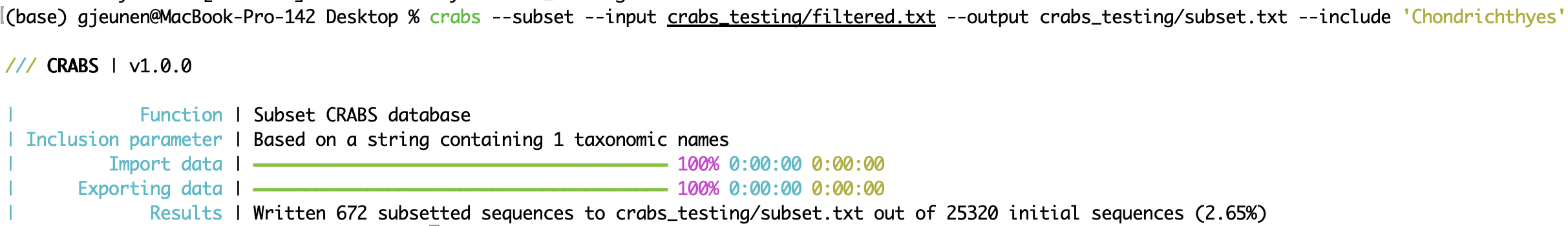
--subset Die dritte und letzte in CRABS integrierte Kurationsmethode besteht darin, die lokale Referenzdatenbank zu unterteilen, um mithilfe der Funktion --subset bestimmte Taxa einzuschließen (Parameter: --include ) oder auszuschließen (Parameter: --exclude ). Diese Funktion ermöglicht das Entfernen von Referenzbarcodes aus taxonomischen Gruppen, die für die Forschungsfrage nicht von Interesse sind. Diese taxonomischen Gruppen könnten aufgrund einer möglichen unspezifischen Amplifikation des Primersatzes in die lokale Referenzdatenbank aufgenommen worden sein. Ein weiterer Anwendungsfall für --subset besteht darin, bekannte fehlerhafte Sequenzen zu entfernen.
Für taxonomische Klassifikatoren, die auf maschinellem Lernen (IDTAXA) oder k-mer-Abstand (SINTAX) basieren, kann es von Vorteil sein, die Referenzdatenbank zu unterteilen, indem nur Taxa einbezogen werden, von denen bekannt ist, dass sie in der Region vorkommen, in der die Proben entnommen wurden, und eng verwandte Arten, von denen bekannt ist, dass sie nicht vorkommen, ausgeschlossen werden in der Region auftreten, um die erhaltene taxonomische Auflösung dieser Klassifikatoren zu erhöhen und verbesserte Taxonomiezuordnungsergebnisse zu erhalten.
Die Eingabe- und Ausgabedateien können mit den Parametern --input bzw. --output angegeben werden. Die Parameter --include und --exclude können entweder eine durch ; getrennte Liste von Taxa aufnehmen ; oder eine TXT-Datei, die einen einzelnen Taxonnamen pro Zeile enthält.
crabs --subset --input crabs_testing/filtered.txt --output crabs_testing/subset.txt --include 'Chondrichthyes'
Sobald die Referenzdatenbank fertiggestellt ist, kann sie in verschiedene Formate exportiert werden, um den Spezifikationen der meisten Softwaretools zur Zuordnung von Taxonomie zu metagenomischen Daten gerecht zu werden. Die Eingabe- und Ausgabedateien können mit den Parametern --input bzw. --output angegeben werden. Ab Version v 1.0.0 integriert CRABS die Formatierung der Referenzdatenbank für sechs verschiedene Klassifikatoren (Parameter: --export-format ), darunter:
--export-format 'sintax' : Der SINTAX-Klassifikator ist in USEARCH und VSEARCH integriert;--export-format 'rdp' : Der RDP-Klassifikator ist ein eigenständiges Programm, das häufig in Mikrobiomstudien verwendet wird.--export-format 'qiime-fasta' und --export-format 'qiime-text' : Kann verwendet werden, um eine taxonomische ID in QIIME und QIIME2 zuzuweisen;--export-format 'dada2-species' und --export-format 'dada2-taxonomy' : Kann verwendet werden, um eine taxonomische ID in DADA2 zuzuweisen;--export-format 'idt-fasta' und --export-format 'idt-text' : Der IDTAXA-Klassifikator ist ein maschineller Lernalgorithmus, der im DECIPHER R-Paket enthalten ist;--export-format 'blast-notax' : Erstellt eine lokale BLAST-Referenzdatenbank für Blastn und Megablast, in der die Ausgabe keine taxonomische ID bereitstellt, sondern die Zugangsnummer auflistet.--export-format 'blast-tax' : Erstellt eine lokale BLAST-Referenzdatenbank für Blastn und Megablast, in der die Ausgabe sowohl die taxonomische ID als auch die Zugangsnummer bietet. crabs --export --input crabs_testing/subset.txt --output crabs_testing/BLAST_TAX_CHONDRICHTHYES --export-format 'blast-tax'

Während der Exportieren der lokalen Referenzdatenbank in ein einzelnes Format (mit Ausnahme der Klassifizierer, in denen die Referenzdatenbank über mehrere Dateien aufgeteilt wird, reicht für die meisten Benutzer ein einfaches für Schleife aus, um den Lokal zu exportieren Referenzdatenbank auf mehrere Formate Wenn Benutzer Ergebnisse zwischen verschiedenen taxonomischen Klassifikatoren vergleichen möchten. Im Folgenden finden Sie unten die lokalen Referenzdatenbank in Sintax-, RDP- und IDTAXA -Formaten.
for format in sintax.fasta rdp.fasta idt-fasta.fasta idt-text.txt; do crabs --export --input crabs_testing/subset.txt --output crabs_testing/chondrichthyes_${format} --export-format ${format%%.*}; done

Once the reference database is finalised, CRABS can run five post-processing functions to explore and provide a summary overview of the local reference database, including (i) --diversity-figure , (ii) --amplicon-length-figure , ( iii) --phylogenetic-tree , (iv) --amplification-efficiency-figure und (v) --completeness-table .
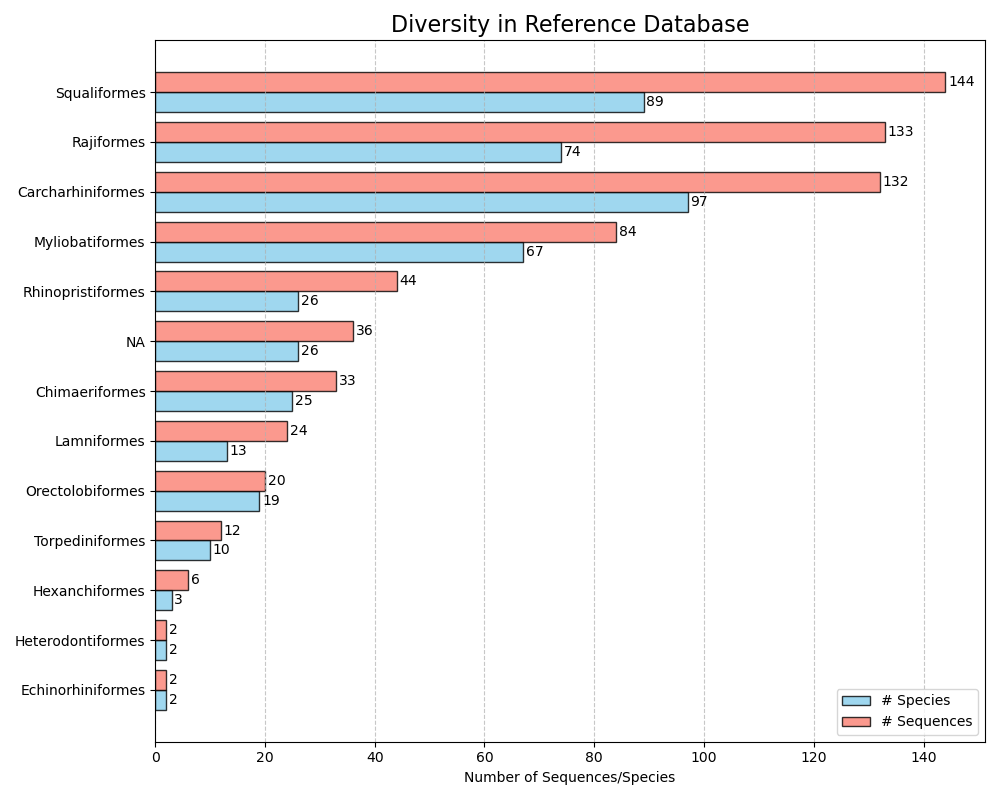
--diversity-figure Die Funktion --diversity-figure Funktion erzeugt ein horizontales Balkendiagramm mit Anzahl der Arten (in blau) und Anzahl der Sequenzen (in orange) pro für jede taxonomische Gruppe in der Referenzdatenbank. Der Benutzer kann den taxonomischen Rang angeben, um die Referenzdatenbank mit dem Parameter --tax-level aufzuteilen. Das Steuerniveau ist die Anzahl des Ranges, in dem sie während der Funktion --import erschien. Zum Beispiel, wenn --ranks 'superkingdom;phylum;class;order;family;genus;species' während der Aufteilung --import verwendet wurde, basierend auf Superkingdom, würde --tax-level 1 , Phylum = --tax-level 2 , erfordern. Class = --tax-level 3 usw. Die Eingabedatei im Crabs-Format kann unter Verwendung des Parameters --input angegeben werden. Die Abbildung im .png -Format wird in die Ausgabedatei geschrieben, die mit dem Parameter --output angegeben werden kann.
crabs --diversity-figure --input crabs_testing/subset.txt --output crabs_testing/diversity-figure.png --tax-level 4
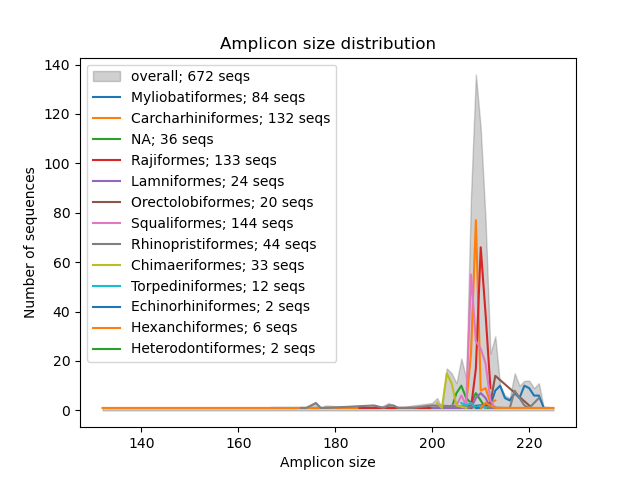
--amplicon-length-figure Die Funktion --amplicon-length-figure Funktion erzeugt ein Liniendiagramm, in dem der Bereich der Amplikonlänge angezeigt wird. Der Gesamtbereich der Amplikonlänge über alle Sequenzen in der Referenzdatenbank wird in einer schattierten grauen Farbe angezeigt, während die Ergebnisse pro taxonomischer Gruppe (Parameter: --tax-level ) durch farbige Linien überlagert werden. Darüber hinaus zeigt die Legende die Anzahl der Sequenzen an, die jeder der taxonomischen Gruppen zugewiesen wurden, und die Gesamtzahl der Sequenzen in der Referenzdatenbank. Die Eingabedatei im Crabs -Format kann unter Verwendung des Parameters --input angegeben werden. Die Abbildung im .png -Format wird in die Ausgabedatei geschrieben, die mit dem Parameter --output angegeben werden kann.
crabs --amplicon-length-figure --input crabs_testing/subset.txt --output crabs_testing/amplicon-length-figure.png --tax-level 4
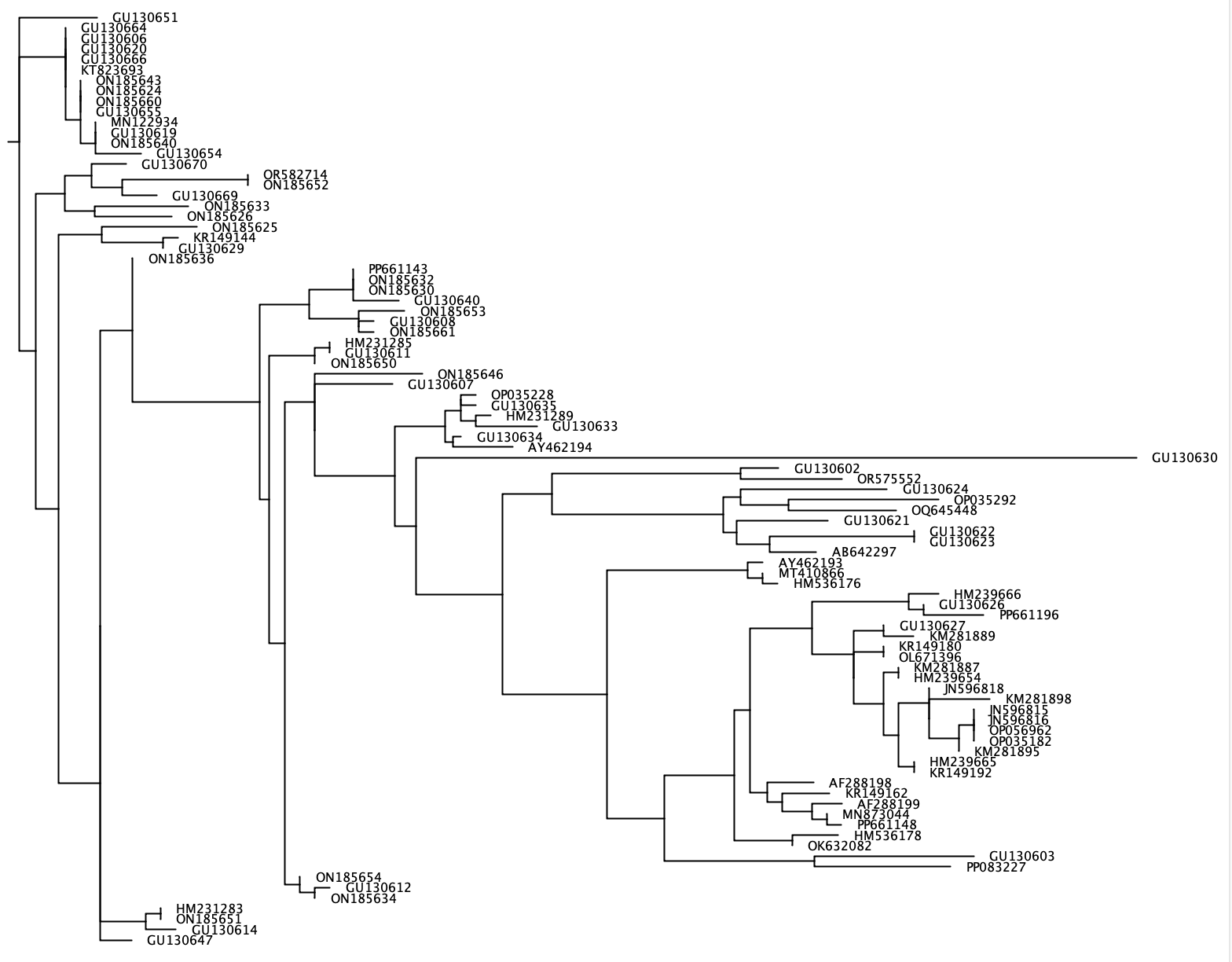
--phylogenetic-tree Die Funktion --phylogenetic-tree Funktion erzeugt einen phylogenetischen Baum für eine Liste von interessierenden Arten. Diese Liste von interessierenden Arten kann unter Verwendung des Parameters --species importiert werden und besteht entweder aus einer Eingangszeichenfolge, die durch + oder eine .txt -Datei mit einem einzelnen Speziesamen in jeder Zeile getrennt ist. Für jede Interessenspezies werden Sequenzen aus der Referenzdatenbank extrahiert, die einen benutzerdefinierten taxonomischen Rang (Parameter: --tax-level ) mit den interessierenden Arten teilen. Krabben erzeugen eine Ausrichtung aller extrahierten Sequenzen unter Verwendung von Clustalw2 V 2.1 und erzeugen einen phylogenetischen Baum mit benachbarten Nachbarn unter Verwendung von Fastree. Dieser phylogenetische Baum im neuen Format wird unter Verwendung des Parameters --output in die Ausgabedatei geschrieben und kann in Softwareprogrammen wie Figree oder Geneious visualisiert werden. Da für jede interessierende Art ein separater phylogenetischer Baum generiert wird, nimmt --output einen generischen Dateinamen ein, während die genaue Ausgabedatei diesen generischen Namen enthält, gefolgt von '_species_name.tree'.
crabs --phylogenetic-tree --input crabs_testing/subset.txt --output crabs_testing/phylo --tax-level 4 --species 'Carcharodon carcharias+Squalus acanthias'
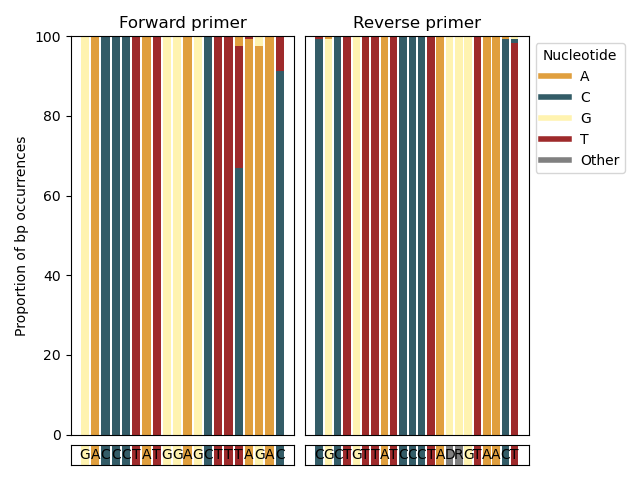
--amplification-efficiency-figure Die Funktion --amplification-efficiency-figure Funktion erzeugt ein Balkendiagramm, wodurch der Anteil des Basispaares Auftreten in den Primer-Bindungsregionen für eine benutzerdefinierte taxonomische Gruppe angezeigt wird, wodurch Orte in den Vorwärts- und Reverse Primer-Binding-Regionen visbyisiert werden Möglicherweise treten in der taxonomischen Interessengruppe auf und beeinflussen möglicherweise die Amplifikationseffizienz. Die Funktion --amplification-efficiency-figure nimmt eine endgültige Krabben-formatierte Referenzdatenbank als Eingabe unter Verwendung des Parameters --amplicons an. Um die Informationen zu den Primer-Bindungsregionen für jede Sequenz in der Eingabedatei zu finden, müssen die ursprünglich heruntergeladenen Sequenzen nach dem Import mit dem Parameter --input bereitgestellt werden. Die Vorwärts- und Reverse Primer -Sequenzen (in 5 ' -3' Richtung) werden unter Verwendung der Parameter --forward und --reverse bereitgestellt. Der Name der taxonomischen Zinsgruppe kann unter Verwendung des Parameters --tax-group bereitgestellt werden und auf jeder taxonomischen Ebene festgelegt werden, die in die Eingabedatei aufgenommen wird. Schließlich wird die Abbildung im .png -Format in die Ausgabedatei geschrieben, die durch den Parameter --output angegeben ist.
crabs --amplification-efficiency-figure --input crabs_testing/merged.txt --amplicons crabs_testing/subset.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT --output crabs_testing/amplification-efficiency.png --tax-group Carcharhiniformes
--completeness-table Die Funktion --completeness-table -Funktion gibt eine tab delimitierte Tabelle (Parameter: --output ) mit Informationen zu einer Liste von Interessensarten aus. Diese Liste von interessierenden Arten kann unter Verwendung des Parameters --species importiert werden und besteht entweder aus einer Eingangszeichenfolge, die durch + oder eine .txt -Datei mit einem einzelnen Speziesamen in jeder Zeile getrennt ist. Für jede Interessenspezies wird eine taxonomische Linie unter Verwendung der Dateien " Namen.dmp " und " Knoten.dmp " generiert, die mit der Funktion --download-taxonomy unter Verwendung der Parameter --names bzw. --nodes heruntergeladen wurden. In der Ausgangstabelle werden 10 Spalten enthält, die folgende Informationen enthalten:
crabs --completeness-table --input crabs_testing/subset.txt --output crabs_testing/completeness.txt --names crabs_testing/names.dmp --nodes crabs_testing/nodes.dmp --species 'Carcharodon carcharias+Squalus acanthias'
crabs --version v 1.0.6 : Bug Fix -> Verbesserte Analyse von kühnen Headern während --import .crabs --version v 1.0.5 : Fehlerbehebung -> Die SEQ -ID bei Erstellung von BLAST -Datenbanken nach Bedarf für die BLAST+ -Software eine Längenbeschränkung hinzugefügt.crabs --version v 1.0.4 : Hinzufügen in Informationen-> Bereitstellung der korrekten Informationen zur Werteingabe für --pairwise-global-alignment --coverage --percent-identity .crabs --version v 1.0.3 : Fehlerfix -> NCBI -Server -Antwort 3 -mal überprüfen, bevor die Analyse abgebrochen wird.crabs --version v 1.0.2 : Fehlerfix -> In der Lage, zu melden, wenn 0 Sequenzen nach der Analyse zurückgegeben werden.crabs --version v 1.0.1 : Fehlerfix -> erfolgreiches Erstellen von NCBI -Abfrage mit dem Parameter --species .


