nucleotide transformer
1.0.0
Willkommen in diesem InstaDeep-Github-Repository, in dem Folgendes vorgestellt wird:
Wir freuen uns, diese Arbeiten als Open Source bereitzustellen und der Community Zugriff auf den Code und die vorab trainierten Gewichte für diese neun Genomik-Sprachmodelle und zwei Segmentierungsmodelle zu ermöglichen. Modelle aus dem nucleotide transformer -Projekt wurden in Zusammenarbeit mit Nvidia und TUM entwickelt und die Modelle wurden auf DGX A100-Knoten auf Cambridge-1 trainiert. Das Modell aus dem Agro- nucleotide transformer -Projekt wurde in Zusammenarbeit mit Google entwickelt und das Modell auf TPU-v4-Beschleunigern trainiert.
Insgesamt liefern unsere Arbeiten neue Erkenntnisse in Bezug auf das Vortraining und die Anwendung sprachbasierter Modelle sowie das Training von Modellen, die sie als Backbone-Encoder verwenden, für die Genomik mit zahlreichen Möglichkeiten ihrer Anwendung in diesem Bereich.
In diesem Repository finden Sie Folgendes:
Im Vergleich zu anderen Ansätzen integrieren unsere Modelle nicht nur Informationen aus einzelnen Referenzgenomen, sondern nutzen DNA-Sequenzen aus über 3.200 verschiedenen menschlichen Genomen sowie 850 Genomen einer breiten Palette von Arten, einschließlich Modell- und Nicht-Modellorganismen. Durch eine solide und umfassende Auswertung zeigen wir, dass diese großen Modelle im Vergleich zu bestehenden Methoden eine äußerst genaue Vorhersage des molekularen Phänotyps ermöglichen.
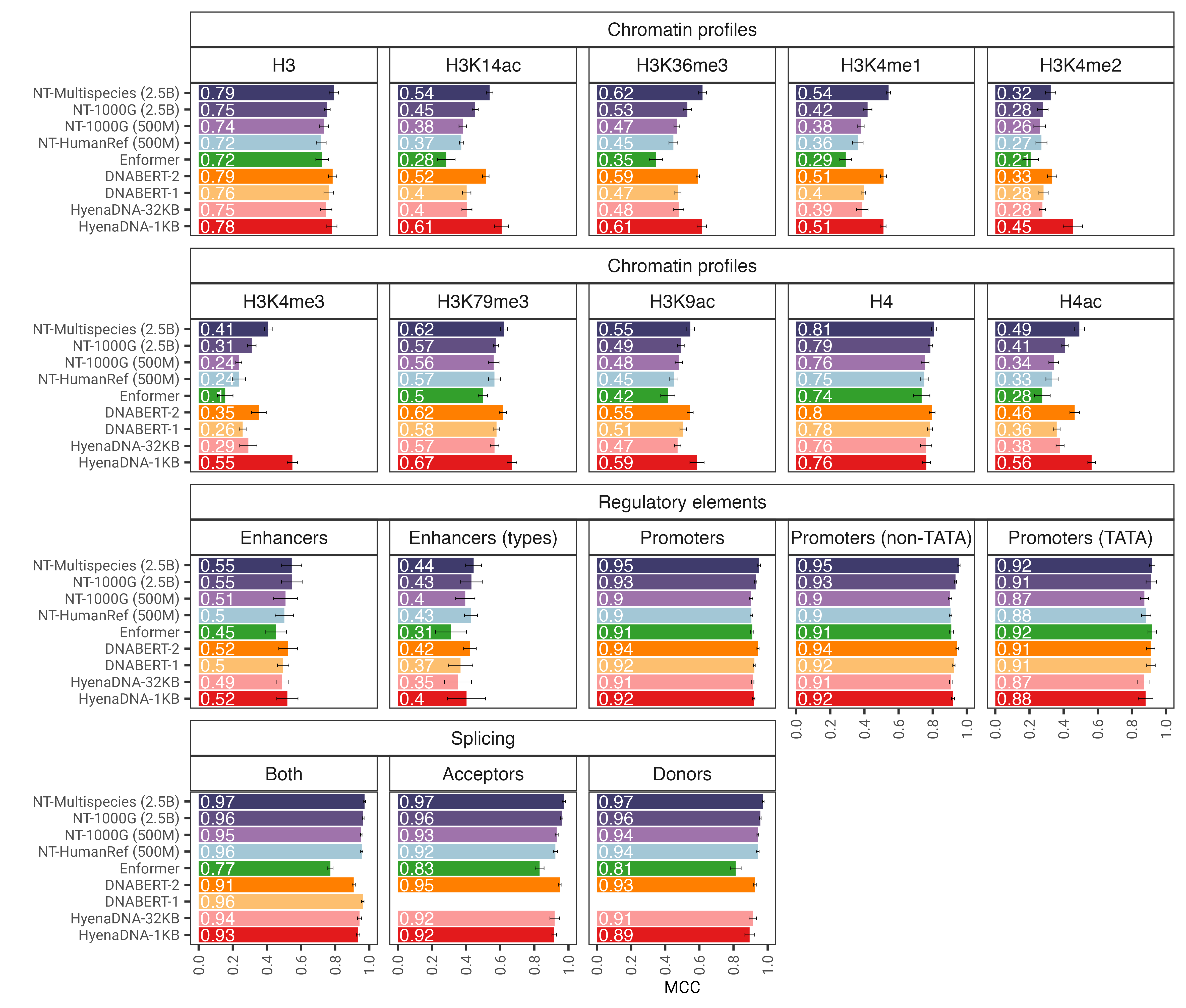
Abb. 1: Das nucleotide transformer sagt nach der Feinabstimmung verschiedene genomische Aufgaben genau voraus. Wir zeigen die Leistungsergebnisse für nachgelagerte Aufgaben für fein abgestimmte Transformatormodelle. Fehlerbalken stellen 2 SDs dar, die aus einer 10-fachen Kreuzvalidierung abgeleitet wurden.
In dieser Arbeit präsentieren wir ein neuartiges grundlegendes großes Sprachmodell, das auf Referenzgenomen von 48 Pflanzenarten trainiert wurde, wobei der Schwerpunkt hauptsächlich auf Nutzpflanzenarten liegt. Wir haben die Leistung von AgroNT bei verschiedenen Vorhersageaufgaben bewertet, die von regulatorischen Merkmalen über die RNA-Verarbeitung bis hin zur Genexpression reichen, und zeigen, dass AgroNT eine Leistung auf dem neuesten Stand der Technik erzielen kann.
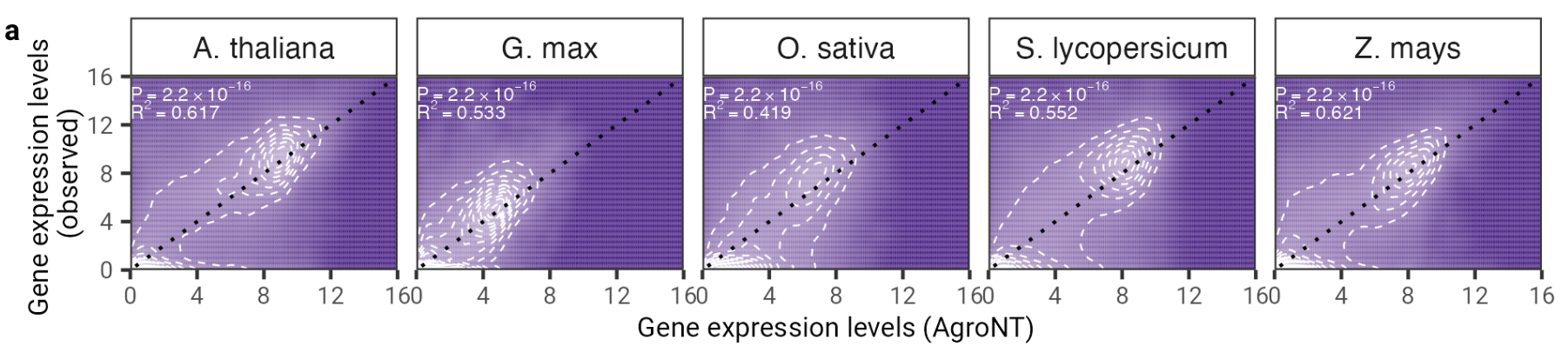
Abb. 2: AgroNT bietet eine Vorhersage der Genexpression für verschiedene Pflanzenarten. Die Vorhersage der Genexpression von Holdout-Genen in allen Geweben korreliert mit den beobachteten Genexpressionsniveaus. Der Bestimmtheitskoeffizient (R 2 ) eines linearen Modells und zugehörige P-Werte zwischen vorhergesagten und beobachteten Werten werden angezeigt.
Um den Code und die vorab trainierten Modelle zu verwenden, gehen Sie einfach wie folgt vor:
pip install . .Anschließend können Sie jedes unserer neun Modelle herunterladen und in nur wenigen Codezeilen die Inferenz durchführen:
import haiku as hk
import jax
import jax . numpy as jnp
from nucleotide_transformer . pretrained import get_pretrained_model
# Get pretrained model
parameters , forward_fn , tokenizer , config = get_pretrained_model (
model_name = "500M_human_ref" ,
embeddings_layers_to_save = ( 20 ,),
max_positions = 32 ,
)
forward_fn = hk . transform ( forward_fn )
# Get data and tokenize it
sequences = [ "ATTCCGATTCCGATTCCG" , "ATTTCTCTCTCTCTCTGAGATCGATCGATCGAT" ]
tokens_ids = [ b [ 1 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens_str = [ b [ 0 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens = jnp . asarray ( tokens_ids , dtype = jnp . int32 )
# Initialize random key
random_key = jax . random . PRNGKey ( 0 )
# Infer
outs = forward_fn . apply ( parameters , random_key , tokens )
# Get embeddings at layer 20
print ( outs [ "embeddings_20" ]. shape )Unterstützte Modellnamen sind:
Sie können unsere Modelle auch ausführen und weiteren Beispielcode in Google Colab finden
Dank Jax läuft der Code sowohl auf der GPU als auch auf der TPU!
Unsere nucleotide transformer v2-Modelle der zweiten Version enthalten eine Reihe von Architekturänderungen, die sich als effizienter erwiesen haben: Anstelle der Verwendung erlernter Positionseinbettungen verwenden wir rotierende Einbettungen, die auf jeder Aufmerksamkeitsebene verwendet werden, und Gated Linear Units mit Swish-Aktivierungen ohne Voreingenommenheit. Diese verbesserten Modelle akzeptieren auch Sequenzen von bis zu 2.048 Token, was zu einem längeren Kontextfenster von 12 kbp führt. Inspiriert von den Chinchilla-Skalierungsgesetzen haben wir unsere NT-v2-Modelle auch auf unserem Multi-Spezies-Datensatz für eine längere Dauer trainiert (300B-Tokens für die 50M- und 100M-Modelle; 1T-Tokens für die 250M- und 500M-Modelle) im Vergleich zu den v1-Modellen (300B-Tokens). für alle vier Modelle).
Die Transformatorschichten sind 1-indiziert, was bedeutet, dass der Aufruf get_pretrained_model mit den Argumenten model_name="500M_human_ref" und embeddings_layers_to_save=(1, 20,) zum Extrahieren von Einbettungen nach der ersten und 20. Transformatorschicht führt. Bei Transformatoren, die den Roberta-LM-Kopf verwenden, ist es üblich, die endgültigen Einbettungen nach der ersten Schichtnorm des LM-Kopfes und nicht nach dem letzten Transformatorblock zu extrahieren. Wenn daher get_pretrained_model mit den folgenden Argumenten embeddings_layers_to_save=(24,) aufgerufen wird, werden die Einbettungen nicht nach der letzten Transformatorschicht, sondern nach der ersten Schichtnorm des LM-Kopfes extrahiert.
SegmentNT-Modelle nutzen einen nucleotide transformer (NT)-Transformator, aus dem wir den Sprachmodellkopf entfernt und durch einen eindimensionalen U-Net-Segmentierungskopf ersetzt haben, um die Position mehrerer Arten von Genomelementen in einer Sequenz mit einer einzigen Nukleotidauflösung vorherzusagen. Wir präsentieren zwei verschiedene Modellvarianten für 14 verschiedene Klassen menschlicher Genomikelemente in Eingabesequenzen von bis zu 30 kb. Dazu gehören Gene (proteinkodierende Gene, lncRNAs, 5'UTR, 3'UTR, Exon, Intron, Spleißakzeptor- und Donorstellen) und regulatorische (PolyA-Signal, gewebeinvariante und gewebespezifische Promotoren und Enhancer sowie CTCF-gebundene Gene). Websites) Elemente. SegmentNT erreicht eine überlegene Leistung gegenüber der hochmodernen U-Net-Segmentierungsarchitektur, profitiert von den vorab trainierten Gewichten von NT und demonstriert eine Zero-Shot-Generalisierung bis zu 50 kbp.
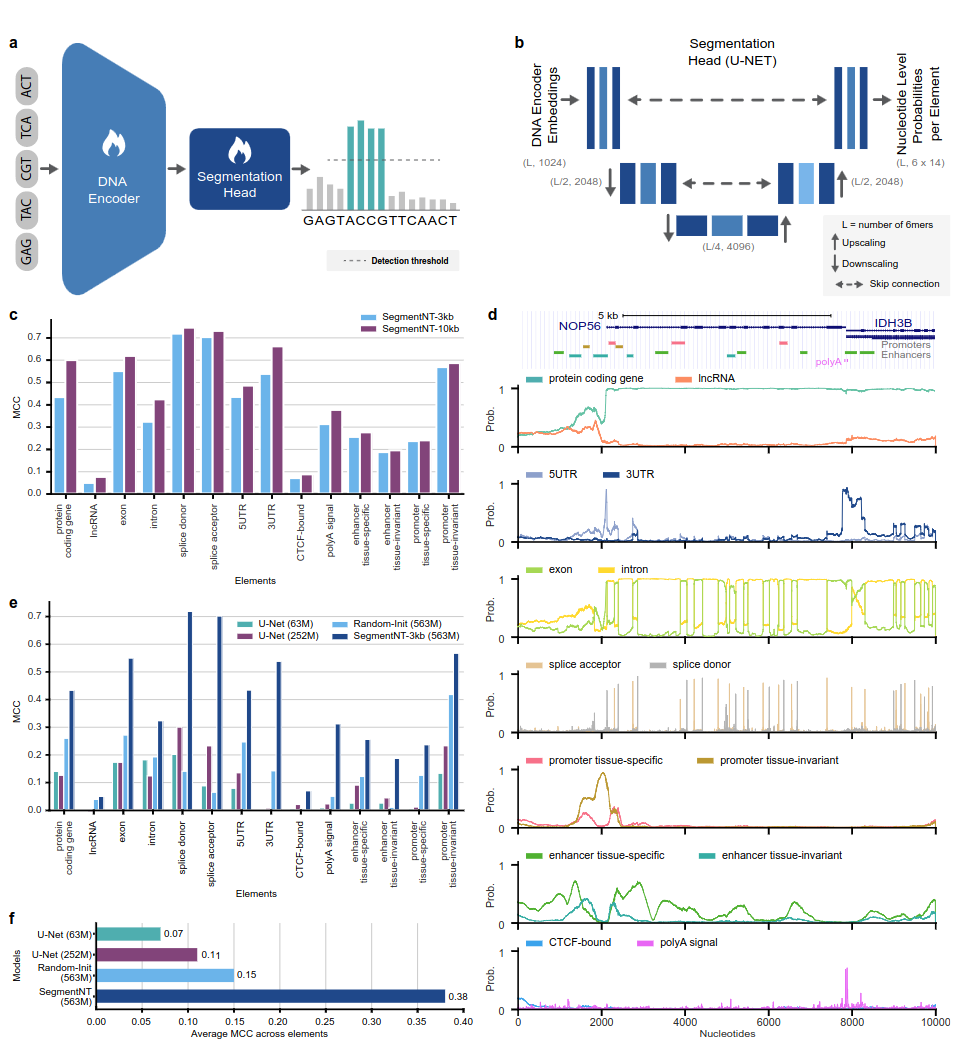
Abb. 1: SegmentNT lokalisiert genomische Elemente mit Nukleotidauflösung.
Um den Code und die vorab trainierten Modelle zu verwenden, gehen Sie einfach wie folgt vor:
pip install . .Anschließend können Sie mit nur wenigen Codezeilen eine Sequenz mit jedem unserer Modelle herunterladen und daraus ableiten:
rescaling factor auf den während des Trainings verwendeten eingestellt. Falls Sie auf Sequenzen zwischen 30 kbp und 50 kbp schließen müssen, stellen Sie sicher, dass Sie das Argument rescaling_factor in der Funktion get_pretrained_segment_nt_model mit dem Wert rescaling_factor = max_num_nucleotides / max_num_tokens_nt übergeben, wobei num_dna_tokens_inference die Anzahl der Tokens bei der Inferenz ist (d. h. 6669 für eine Sequenz von). 40008 Basenpaare) und max_num_tokens_nt ist die maximale Anzahl von Tokens, auf denen der Backbone-Nukleotid-Transformer trainiert wurde, also 2048 .
? Das Notizbuch examples/inference_segment_nt.ipynb zeigt, wie man auf eine 50-kb-Sequenz schließt und die Wahrscheinlichkeiten darstellt, um die Abbildung 3 des Papiers zu reproduzieren.
? Die SegmentNT-Modelle verarbeiten kein „N“ in der Eingabesequenz, da jedes Nukleotid als 6-mere tokenisiert werden muss, was nicht der Fall sein kann, wenn Sequenzen verwendet werden, die ein oder mehrere „N“-Basenpaare enthalten.
import haiku as hk
import jax
import jax . numpy as jnp
from nucleotide_transformer . pretrained import get_pretrained_segment_nt_model
# Initialize CPU as default JAX device. This makes the code robust to memory leakage on
# the devices.
jax . config . update ( "jax_platform_name" , "cpu" )
backend = "cpu"
devices = jax . devices ( backend )
num_devices = len ( devices )
print ( f"Devices found: { devices } " )
# The number of DNA tokens (excluding the CLS token prepended) needs to be dividible by
# 2 to the power of the number of downsampling block, i.e 4.
max_num_nucleotides = 8
assert max_num_nucleotides % 4 == 0 , (
"The number of DNA tokens (excluding the CLS token prepended) needs to be dividible by"
"2 to the power of the number of downsampling block, i.e 4." )
parameters , forward_fn , tokenizer , config = get_pretrained_segment_nt_model (
model_name = "segment_nt" ,
embeddings_layers_to_save = ( 29 ,),
attention_maps_to_save = (( 1 , 4 ), ( 7 , 10 )),
max_positions = max_num_nucleotides + 1 ,
)
forward_fn = hk . transform ( forward_fn )
apply_fn = jax . pmap ( forward_fn . apply , devices = devices , donate_argnums = ( 0 ,))
# Get data and tokenize it
sequences = [ "ATTCCGATTCCGATTCCAACGGATTATTCCGATTAACCGATTCCAATT" , "ATTTCTCTCTCTCTCTGAGATCGATGATTTCTCTCTCATCGAACTATG" ]
tokens_ids = [ b [ 1 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens_str = [ b [ 0 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens = jnp . asarray ( tokens_ids , dtype = jnp . int32 )
random_key = jax . random . PRNGKey ( seed = 0 )
keys = jax . device_put_replicated ( random_key , devices = devices )
parameters = jax . device_put_replicated ( parameters , devices = devices )
tokens = jax . device_put_replicated ( tokens , devices = devices )
# Infer on the sequence
outs = apply_fn ( parameters , keys , tokens )
# Obtain the logits over the genomic features
logits = outs [ "logits" ]
# Transform them in probabilities
probabilities = jnp . asarray ( jax . nn . softmax ( logits , axis = - 1 ))[..., - 1 ]
print ( f"Probabilities shape: { probabilities . shape } " )
print ( f"Features inferred: { config . features } " )
# Get probabilities associated with intron
idx_intron = config . features . index ( "intron" )
probabilities_intron = probabilities [..., idx_intron ]
print ( f"Intron probabilities shape: { probabilities_intron . shape } " )Unterstützte Modellnamen sind:
Dank Jax läuft der Code sowohl auf der GPU als auch auf der TPU!
Die Modelle werden auf Sequenzen mit einer Länge von bis zu 1000 Token trainiert, einschließlich des <CLS>-Tokens, das automatisch am Anfang der Sequenz vorangestellt wird. Der Tokenizer beginnt mit der Tokenisierung von links nach rechts, indem er die Buchstaben „A“, „C“, „G“ und „T“ in 6-meren gruppiert. Der Buchstabe „N“ ist so gewählt, dass er nicht innerhalb der k-mers gruppiert wird. Daher werden die Nukleotide ohne Gruppierung tokenisiert, wenn der Tokenizer auf ein „N“ trifft oder wenn die Anzahl der Nukleotide in der Sequenz kein Vielfaches von 6 ist ihnen. Beispiele sind unten aufgeführt:
dna_sequence_1 = "ACGTGTACGTGCACGGACGACTAGTCAGCA"
tokenized_dna_sequence_1 = [ < CLS > , < ACGTGT > , < ACGTGC > , < ACGGAC > , < GACTAG > , < TCAGCA > ]
dna_sequence_2 = "ACGTGTACNTGCACGGANCGACTAGTCTGA"
tokenized_dna_sequence_2 = [ < CLS > , < ACGTGT > , < A > , < C > , < N > , < TGCACG > , < G > , < A > , < N > , < CGACTA > , < GTCTGA > ]Alle v1- und v2-Transformatoren können daher Sequenzen mit bis zu 5994 bzw. 12282 Nukleotiden annehmen, wenn darin kein „N“ enthalten ist.
Die in diesem Repository präsentierte Sammlung von Modellen ist hier in den Huggingface Spaces von Instadeep verfügbar: The nucleotide transformer Space und Agro nucleotide transformer Space!
Wir danken Maša Roller sowie den Mitgliedern des Rostlab, insbesondere Tobias Olenyi, Ivan Koludarov und Burkhard Rost, für konstruktive Diskussionen, die dazu beigetragen haben, interessante Forschungsrichtungen zu identifizieren. Darüber hinaus danken wir allen, die experimentelle Daten in öffentlichen Datenbanken hinterlegen, denen, die diese Datenbanken pflegen, und denen, die analytische und prädiktive Methoden frei verfügbar machen. Wir danken auch dem Jax-Entwicklungsteam.
Wenn Sie dieses Repository für Ihre Arbeit nützlich finden, fügen Sie bitte einem unserer zugehörigen Artikel ein entsprechendes Zitat hinzu:
Das nucleotide transformer Papier:
@article { dalla2023nucleotide ,
title = { The nucleotide transformer : Building and Evaluating Robust Foundation Models for Human Genomics } ,
author = { Dalla-Torre, Hugo and Gonzalez, Liam and Mendoza Revilla, Javier and Lopez Carranza, Nicolas and Henryk Grywaczewski, Adam and Oteri, Francesco and Dallago, Christian and Trop, Evan and Sirelkhatim, Hassan and Richard, Guillaume and others } ,
journal = { bioRxiv } ,
pages = { 2023--01 } ,
year = { 2023 } ,
publisher = { Cold Spring Harbor Laboratory }
}Agro- nucleotide transformer :
@article { mendoza2024foundational ,
title = { A foundational large language model for edible plant genomes } ,
author = { Mendoza-Revilla, Javier and Trop, Evan and Gonzalez, Liam and Roller, Ma{v{s}}a and Dalla-Torre, Hugo and de Almeida, Bernardo P and Richard, Guillaume and Caton, Jonathan and Lopez Carranza, Nicolas and Skwark, Marcin and others } ,
journal = { Communications Biology } ,
volume = { 7 } ,
number = { 1 } ,
pages = { 835 } ,
year = { 2024 } ,
publisher = { Nature Publishing Group UK London }
}SegmentNT-Papier
@article { de2024segmentnt ,
title = { SegmentNT: annotating the genome at single-nucleotide resolution with DNA foundation models } ,
author = { de Almeida, Bernardo P and Dalla-Torre, Hugo and Richard, Guillaume and Blum, Christopher and Hexemer, Lorenz and Gelard, Maxence and Pandey, Priyanka and Laurent, Stefan and Laterre, Alexandre and Lang, Maren and others } ,
journal = { bioRxiv } ,
pages = { 2024--03 } ,
year = { 2024 } ,
publisher = { Cold Spring Harbor Laboratory }
}Wenn Sie Fragen oder Feedback zum Code und den Modellen haben, können Sie sich gerne an uns wenden.
Vielen Dank für Ihr Interesse an unserer Arbeit!


