BRAKER
v3.0.8
Hier finden Sie eine Aufzeichnung der ersten BGA23 -Workshop -Sitzung über Braker. Wenn Sie das Lernen durch Ansehen von Videos für Sie einfach haben, sollten Sie sich das ansehen: https://www.youtube.com/watch?v=uxtkj4mukyg
Braker3 ist jetzt in https://usegalaxy.eu/
Tsebra & Braker3 verwandt:
Braker & Augustus verwandt:
Genemark verwandt:
Mark Borodovsky, Georgia Tech, USA, [email protected]
Tomas Bruna, Joint Genome Institute, USA, [email protected]
Alexandre Lomsazde, Georgia Tech, USA, [email protected]
[A] Universität Greifwald, Institut für Mathematik und Informatik, Walther-Rathenau-Str. 47, 17489 Greifswald, Deutschland
[b] Universität Greifswald, Zentrum für funktionelle Genomik von Mikroben, Felix-Haudorff-Str. 8, 17489 Greifswald, Deutschland
[C] Gemeinsame Georgia Tech und Emory University Wallace H Coulter Abteilung für Biomedizinische Ingenieurwesen, 30332 Atlanta, USA
[D] School of Computational Science and Engineering, 30332 Atlanta, USA
[E] Moskauer Institut für Physik und Technologie, Moskauer Region 141701, Dolgopudny, Russland
![Braker2-Team-2 [Fig. 10]](https://images.downcodes.com/uploads/20250214/img_67aee79a0cb7530.png)
![Braker2-Team-1 [Fig11]](https://images.downcodes.com/uploads/20250214/img_67aee79a0d1eb31.png)
![Braker2-Team-3 [Abb. 12]](https://images.downcodes.com/uploads/20250214/img_67aee79a0da9c32.png)
![Braker2-Team-4 [Abb. 13]](https://images.downcodes.com/uploads/20250214/img_67aee79a0e49933.png)
Abbildung 1: Aktuelle Braker -Autoren von links nach rechts: Mario Stanke, Alexandre Lomsadze, Katharina J. Hoff, Tomas Bruna, Lars Gabriel und Mark Borodovsky. Wir erkennen an, dass eine größere Gemeinschaft von Wissenschaftlern zum Braker -Code beigetragen hat (z. B. über Pull -Anfragen).
Die Entwicklung von Braker1, Braker2 und Braker3 wurde von den National Institutes of Health (NIH) [GM128145 an MB und MS] unterstützt. Die Entwicklung von Braker3 wurde teilweise durch Projektdatenkompetenz finanziert, die KJH und MS von der Regierung von Mecklenburg-Vorpommern, Deutschland, gewährt wurden.
Der Transkript-Selektor für Braker (Tsebra) ist unter https://github.com/gaius-augustus/tsebra erhältlich.
Genemark-etp, einer der Gen-Finder im Kern von Braker, ist unter https://github.com/gatech-genemark/genemark-etp erhältlich.
Augustus, der zweite Genfinder im Kern von Braker, ist unter https://github.com/gaius-augustus/augustus erhältlich.
Galba, ein Braker-Pipeline-Spin-off für die Verwendung von Miniprot oder Genomethreader zur Generierung von Trainingsgenen, ist unter https://github.com/gaius-augustus/galba verfügbar.
Die schnell wachsende Anzahl sequenzierter Genome erfordert vollständig automatisierte Methoden für eine genaue Annotation der Genstruktur. Vor diesem Hintergrund haben wir Braker1 R1 R0 , eine Kombination aus Genemark-ET -R2 und Augustus R3, R4 , entwickelt, die genomische und rNA-seq-Daten verwendet, um automatisch vollständige Annotationen der Genstruktur im neuartigen Genom zu erzeugen.
Die Qualität von RNA-Seq-Daten, die für die Annotation eines neuartigen Genoms verfügbar sind, ist jedoch variabel, und in einigen Fällen sind RNA-Seq-Daten überhaupt nicht verfügbar.
Braker2 ist eine Erweiterung von Braker1, die eine vollständig automatisierte Schulung der Genvorhersagewerkzeuge Genemark-ES/EP/ETP R14, R15, R17, F1 und Augustus ermöglicht Extrinsische Nachweise aus Informationen über RNA-Seq und Proteinhomologie in die Vorhersage .
Im Gegensatz zu anderen verfügbaren Methoden, die sich auf Informationen zur Proteinhomologie beruhen, erreicht Braker2 auch ohne die Annotation von sehr eng verwandten Arten und in Abwesenheit von RNA-Seq-Daten eine hohe Genvorhersagegenauigkeit.
Braker3 ist die neueste Pipeline in der Braker Suite. Es ermöglicht die Verwendung von RNA-Seq- und Proteindaten in einer vollständig automatisierten Pipeline, um sehr zuverlässige Gene mit Genemark-Etp und Augustus zu trainieren und vorherzusagen. Das Ergebnis der Pipeline ist der kombinierte Gensatz beider Genvorhersagewerkzeuge, die nur Gene mit sehr hoher Unterstützung durch extrinsische Beweise enthält.
In diesem Benutzerhandbuch verweisen wir auf Braker1, Braker2 und Braker3 einfach als Braker, da sie von demselben Skript ( braker.pl ) ausgeführt werden.
Verwenden Sie eine hochwertige Genomanordnung. Wenn Sie eine große Anzahl sehr kurzer Gerüste in Ihrer Genomanordnung haben, erhöhen diese kurzen Gerüste wahrscheinlich die Laufzeit dramatisch, erhöhen jedoch die Vorhersagegenauigkeit nicht.
Verwenden Sie einfache Gerüstnamen in der Genomdatei (z. B. >contig1 funktioniert besser als >contig1my custom species namesome putative function /more/information/ and lots of special characters %&!*(){} ). Machen Sie die Gerüstnamen in all Ihren Fasta -Dateien einfach, bevor Sie ein Ausrichtungsprogramm ausführen.
Um Gene in einem neuen Genom genau vorherzusagen, sollte das Genom für Wiederholungen maskiert werden. Dies vermeidet die Vorhersage falsch positiver Genstrukturen in sich wiederholenden und niedrig komplexen Regionen. Wiederholungsmaskierung ist auch für die Abbildung von RNA-Seq-Daten in ein Genom mit einigen Werkzeugen (andere RNA-Seq-Mapper wie Hisat2, Ignorieren Sie Maskierungsinformationen) von wesentlicher Bedeutung. Im Falle von Genemark-ES/et/EP/ETP und Augustus führt Softmasking (dh das Einfügen von Wiederholungsregionen in untere Fallbuchstaben und alle anderen Regionen in obere Fallbuchstaben) zu besseren Ergebnissen als Hardmasking (dh Ersetzung von Buchstaben in repetitiven Regionen durch den Brief N für unbekanntes Nukleotid).
Viele Genome haben Genstrukturen, die genau mit Standardparametern von Genemark-ES/EP/ETP und Augustus innerhalb des Brakers vorhergesagt werden. Einige Genome haben jedoch kladenspezifische Merkmale, ein spezielles Zweigpunktmodell in Pilzen oder nicht standardmäßige Spleiß-Site-Muster. Bitte lesen Sie den Abschnitt Optionen [Optionen], um festzustellen, ob eine der benutzerdefinierten Optionen die Genom -Vorhersage der Genvorhersage im Genom Ihrer Zielarten verbessern kann.
Überprüfen Sie immer die Vorhersageergebnisse der Genvorhersage, bevor Sie weiter verwendet werden! Sie können zB einen Genombrowser verwenden, um die Genmodelle im Kontext mit extrinsischen Evidenzdaten zu untersuchen. Braker unterstützt die Erzeugung von Track -Daten -Hubs für den UCSC -Genombrowser mit Makehub für diesen Zweck.
Brakern enthält hauptsächlich semi-nicht überträgte, extrinsische Beweisdaten (RNA-Seq- und/oder Proteinspeisen-Ausrichtungsinformationen), unterstützte das Training von Genemark-ES/EP/ETP [F1] und anschließendes Training von Augustus mit Integration von extrinsischen Beweisen in die Schluss Genvorhersageschritt. In Braker gibt es jetzt jedoch eine Reihe zusätzlicher Pipelines. Im Folgenden geben wir einen Überblick über mögliche Eingabedateien und Pipelines:
![Braker2-Main-A [Abb. 1]](https://images.downcodes.com/uploads/20250214/img_67aee79a0eaf534.png)
Abbildung 2: Braker-Pipeline A: Training Genemark-ES nur für Genomdaten; Ab initio Gene Vorhersage mit Augustus
![Braker2-Main-B [Abb. 2]](https://images.downcodes.com/uploads/20250214/img_67aee79a0f13f35.png)
Abbildung 3: Braker-Pipeline B: Training Genemark-et unterstützt durch RNA-Seq Spleiß-Ausrichtungsinformationen, Vorhersage mit Augustus mit den gleichen Spleißausrichtungsinformationen.
![Braker2-Main-C [Abb. 3]](https://images.downcodes.com/uploads/20250214/img_67aee79a0fa1036.png)
Abbildung 4: Braker-Pipeline C: Training Genemark-ep+ auf Proteinspeigeblöste Ausrichtung, Start und Stoppinformationen, Vorhersage mit Augustus mit denselben Informationen, zusätzlich verkettete CDSPart-Hinweise. Hier verwendete Proteine können von jeder evolutionären Entfernung zum Zielorganismus sein.
![Braker3-Main-A [Abb4]](https://images.downcodes.com/uploads/20250214/img_67aee79a1010b37.png)
Abbildung 5: Braker-Pipeline D: Download und Ausrichtung von RNA-Seq-Sätzen für die Zielarten bei Bedarf. Schulung von Genemark-ETP, unterstützt durch die RNA-Seq-Alignments und eine große Proteindatenbank (Proteine können jede evolutionäre Entfernung sein). Anschließend das Augustus-Training und die Vorhersage der gleichen extrinsischen Informationen zusammen mit den Genemark-ETP-Ergebnissen. Die endgültige Vorhersage ist die Tsebra-Kombination der Ergebnisse Augustus und Genemark-ETP.
Wir sind uns bewusst, dass die "manuelle" Installation von Braker3 und all seiner Abhängigkeiten langweilig und ohne Wurzelberechtigungen wirklich herausfordernd ist. Daher bieten wir einen Docker -Container an, der entwickelt wurde, um mit Singularität betrieben zu werden. Alle Informationen zu diesem Container finden Sie unter https://hub.docker.com/r/teambraker/braker3
Kurz gesagt, bauen Sie es wie folgt auf:
singularity build braker3.sif docker://teambraker/braker3:latest
Ausführen mit:
singularity exec braker3.sif braker.pl
Test mit:
singularity exec -B $PWD:$PWD braker3.sif cp /opt/BRAKER/example/singularity-tests/test1.sh .
singularity exec -B $PWD:$PWD braker3.sif cp /opt/BRAKER/example/singularity-tests/test2.sh .
singularity exec -B $PWD:$PWD braker3.sif cp /opt/BRAKER/example/singularity-tests/test3.sh .
export BRAKER_SIF=/your/path/to/braker3.sif # may need to modify
bash test1.sh
bash test2.sh
bash test3.sh
Nur wenige Benutzer möchten ihre Analyse in Docker ausführen (da Root -Berechtigungen erforderlich sind). Wenn dies jedoch Ihr Ziel ist, können Sie den Container wie folgt laufen und testen
sudo docker run --user 1000:100 --rm -it teambraker/braker3:latest bash
bash /opt/BRAKER/example/docker-tests/test1.sh # BRAKER1
bash /opt/BRAKER/example/docker-tests/test2.sh # BRAKER2
bash /opt/BRAKER/example/docker-tests/test3.sh # BRAKER3
Viel Glück ;-)
$PATH -Variablen verweilen, zu unvorhergesehenen Interferenzen führen und Programmfehler verursachen. Bitte bewegen Sie alle älteren Genemark -Versionen aus Ihrem $PATH (auch in der Genemark in ProtHint/dependencies ).
Zum Zeitpunkt der Veröffentlichung wurde diese Braker -Version getestet mit:
Augustus 3.5.0 F2
Genemark-etp (Quelle siehe Dockerfile)
BAMTOOLS 2.5.1 R5
SAMTOOLS 1.7-4-G93586 R6
Spaln 2.3.3d R8, R9, R10
NCBI Blast+ 2.2.31+ R12, R13
Diamant 0.9.24
Cdbfasta 0,99
CDByank 0,981
Gushr 1.0.0
SRA Toolkit 3.00 R14
HISAT2 2.2.1 R15
Bedtools 2.30 R16
StringTie2 2.2.1 R17
GFFREAD 0.12.7 R18
complasm 0.2.5 R27
Running Braker erfordert ein Linux-System mit bash und Perl. Darüber hinaus benötigen Braker die folgenden CPAN-PERL-Module, die installiert werden müssen:
File::Spec::Functions
Hash::Merge
List::Util
MCE::Mutex
Module::Load::Conditional
Parallel::ForkManager
POSIX
Scalar::Util::Numeric
YAML
Math::Utils
File::HomeDir
Für Genemark-ETP, verwendet, wenn Protein und RNA-Seq geliefert werden:
YAML::XSData::DumperThread::Queuethreads Installieren Sie beispielsweise bei Ubuntu die Module mit Cpanminus F4 : sudo cpanm Module::Name , z. B. sudo cpanm Hash::Merge .
Braker verwendet auch ein Perl -Modul helpMod_braker.pm , das auf CPAN nicht verfügbar ist. Dieses Modul ist Teil der Braker -Version und erfordert keine separate Installation.
Wenn Sie keine Root -Berechtigungen auf der Linux -Maschine haben, stellen Sie wie folgt ein Anaconda (https://www.anaconda.com/distribution/) ein.
wget https://repo.anaconda.com/archive/Anaconda3-2018.12-Linux-x86_64.sh
bash bin/Anaconda3-2018.12-Linux-x86_64.sh # do not install VS (needs root privileges)
conda install -c anaconda perl
conda install -c anaconda biopython
conda install -c bioconda perl-app-cpanminus
conda install -c bioconda perl-file-spec
conda install -c bioconda perl-hash-merge
conda install -c bioconda perl-list-util
conda install -c bioconda perl-module-load-conditional
conda install -c bioconda perl-posix
conda install -c bioconda perl-file-homedir
conda install -c bioconda perl-parallel-forkmanager
conda install -c bioconda perl-scalar-util-numeric
conda install -c bioconda perl-yaml
conda install -c bioconda perl-class-data-inheritable
conda install -c bioconda perl-exception-class
conda install -c bioconda perl-test-pod
conda install -c bioconda perl-file-which # skip if you are not comparing to reference annotation
conda install -c bioconda perl-mce
conda install -c bioconda perl-threaded
conda install -c bioconda perl-list-util
conda install -c bioconda perl-math-utils
conda install -c bioconda cdbtools
conda install -c eumetsat perl-yaml-xs
conda install -c bioconda perl-data-dumper
Installieren Sie anschließend Braker und andere Software "wie üblich", während Sie in Ihrer Conda -Umgebung sind. Hinweis: Es gibt ein Bioconda Braker -Paket und ein Bioconda Augustus -Paket. Sie arbeiten. Sie sind jedoch normalerweise hinter dem Entwicklungscode beider Tools auf GitHub zurück. Wir empfehlen daher die manuelle Installation und Verwendung der neuesten Quellen.
Braker ist eine Sammlung von Perl- und Python -Skripten und einem Perl -Modul. Das Hauptskript, das aufgerufen wird, um Braker auszuführen, ist braker.pl . Zusätzliche Perl- und Python -Komponenten sind:
align2hints.pl
filterGenemark.pl
filterIntronsFindStrand.pl
startAlign.pl
helpMod_braker.pm
findGenesInIntrons.pl
downsample_traingenes.pl
ensure_n_training_genes.py
get_gc_content.py
get_etp_hints.py
Alle Skripte (Dateien, die mit *.pl und *.py ) enden, die Teil von Braker sind, müssen ausführbar sein, um Braker auszuführen. Dies sollte bereits der Fall sein, wenn Sie Braker von GitHub herunterladen. Die Ausführbarkeit kann überschrieben werden, wenn Sie z. B. Braker auf einem USB-Stick auf einen anderen Computer übertragen. Um zu überprüfen, ob die erforderlichen Dateien ausführbar sind, führen Sie den folgenden Befehl im Verzeichnis aus, der Braker Perl -Skripte enthält:
ls -l *.pl *.py
Die Ausgabe sollte dem ähnlich sein:
-rwxr-xr-x 1 katharina katharina 18191 Mai 7 10:25 align2hints.pl
-rwxr-xr-x 1 katharina katharina 6090 Feb 19 09:35 braker_cleanup.pl
-rwxr-xr-x 1 katharina katharina 408782 Aug 17 18:24 braker.pl
-rwxr-xr-x 1 katharina katharina 5024 Mai 7 10:25 downsample_traingenes.pl
-rwxr-xr-x 1 katharina katharina 5024 Mai 7 10:23 ensure_n_training_genes.py
-rwxr-xr-x 1 katharina katharina 4542 Apr 3 2019 filter_augustus_gff.pl
-rwxr-xr-x 1 katharina katharina 30453 Mai 7 10:25 filterGenemark.pl
-rwxr-xr-x 1 katharina katharina 5754 Mai 7 10:25 filterIntronsFindStrand.pl
-rwxr-xr-x 1 katharina katharina 7765 Mai 7 10:25 findGenesInIntrons.pl
-rwxr-xr-x 1 katharina katharina 1664 Feb 12 2019 gatech_pmp2hints.pl
-rwxr-xr-x 1 katharina katharina 2250 Jan 9 13:55 log_reg_prothints.pl
-rwxr-xr-x 1 katharina katharina 4679 Jan 9 13:55 merge_transcript_sets.pl
-rwxr-xr-x 1 katharina katharina 41674 Mai 7 10:25 startAlign.pl
Es ist wichtig, dass das x in -rwxr-xr-x für jedes Skript vorhanden ist. Wenn dies nicht der Fall ist, laufen Sie
`chmod a+x *.pl *.py`
um Dateiattribute zu ändern.
Möglicherweise finden Sie es hilfreich, das Verzeichnis hinzuzufügen, in dem sich Braker Perl -Skripte Ihrer $PATH -Umgebungsvariablen befinden. Geben Sie für eine einzige Bash -Sitzung ein:
PATH=/your_path_to_braker/:$PATH
export PATH
Fügen Sie die obigen Zeilen zu einem Startskript (zB ~/.bashrc ) hinzu, um diese $PATH -Änderung für alle Bash -Sitzungen zur Verfügung zu stellen.
Braker fordert verschiedene Bioinformatik -Softwaretools auf, die nicht Teil von Braker sind. Einige Tools sind obligatorisch, dh Braker wird überhaupt nicht ausgeführt, wenn diese Tools nicht in Ihrem System vorhanden sind. Andere Tools sind optional. Bitte installieren Sie alle Tools, die zum Ausführen von Brakern im Modus Ihrer Wahl erforderlich sind.
Download Genemark-etp F1 von http://github.com/gatech-genemark/genemark-etp oder https://topaz.gatech.edu/genemark/etp.for_braker.tar.gz. Entpacken und installieren Sie Genemark-ETP, wie in der README Datei von Genemark-ETP beschrieben.
Wenn der Braker bereits in Ihrer $PATH -Variablen enthalten ist, erraten Braker den Standort von gmes_petap.pl oder gmetp.pl automatisch. Andernfalls kann Braker geneMark-es/et/eP/ETP-ausführbare Produkte entweder in einer Umgebungsvariablen GENEMARK_PATH oder durch die Übernahme eines Befehlszeilenarguments finden ( --GENEMARK_PATH=/your_path_to_GeneMark_executables/ ).
Um die Umgebungsvariable für Ihre aktuelle Bash -Sitzung festzulegen, geben Sie ein:
export GENEMARK_PATH=/your_path_to_GeneMark_executables/
Fügen Sie die obigen Zeilen einem Startskript (z. B. ~/.bashrc ) hinzu, um es allen Bash -Sitzungen zur Verfügung zu stellen.
Perl-Skripte innerhalb von Genemark-ES/ET/EP/ETP werden mit Standard-Perl-Speicherort unter /usr/bin/perl konfiguriert.
Wenn Sie Genemark-ES/et/eP/ETP in einer Anaconda-Umgebung ausführen (oder Perl aus der $PATH Variablen aus einem anderen Grund verwenden möchten), ändern Sie den Shebang aller Genemark-ES/ET/EP/ETP-Skripte mit Der folgende Befehl im Ordner Genemark-ES/ET/EP/ETP:
perl change_path_in_perl_scripts.pl "/usr/bin/env perl"
Sie können überprüfen, ob Genemark-ES/ET/EP ordnungsgemäß installiert wird, indem Sie das Verzeichnis von Genemark-e check_install.bash ausführen und Beispiele im GeneMark-E-tests ausführen.
Genemark-ETP ist nach unten kompatibel, dh die Funktionalität von Genemark-EP und Genemark-Et in Brakern.
Laden Sie Augustus von seiner Master-Filiale unter https://github.com/gaius-augustus/augustus herunter. Augustus auspacken und Augustus laut Augustus README.TXT installieren. Verwenden Sie keine veralteten Augustus -Versionen aus anderen Quellen, z. B. Debian -Paket oder Bioconda -Paket! Braker hängt insbesondere von einem aktuellen Augustus/Skript-Verzeichnis ab, und andere Quellen sind oft zurückgeblieben.
Sie sollten Augustus auf Ihrem eigenen System zusammenstellen, um Probleme mit Versionen von Bibliotheken zu vermeiden, die von Augustus verwendet werden. Kompilierungsanweisungen finden Sie in der Augustus README.TXT -Datei ( Augustus/README.txt ).
Augustus besteht aus augustus , dem Genvorhersage -Tool, zusätzlichen C ++ - Tools in Augustus/auxprogs und Perl -Skripten in Augustus/scripts . Perl -Skripte müssen ausführbar sein (siehe Anweisungen in Braker -Komponenten der Abschnitt.
Das C ++ TOOL bam2hints ist ein wesentlicher Bestandteil von Braker, wenn er mit RNA-Seq ausgeführt wird. Quellen befinden sich in Augustus/auxprogs/bam2hints . Stellen Sie sicher, dass Sie bam2hints in Ihrem System kompilieren (es sollte automatisch zusammengestellt werden, wenn Augustus kompiliert wird. Bei Problemen mit bam2hints jedoch lesen Sie bitte die Anweisungen zur Fehlerbehebung in Augustus/auxprogs/bam2hints/README ).
Da Braker eine Pipeline ist, die Augustus trainiert, schreibt der IE speziesspezifische Parameterdateien. Braker muss zu Zugriff auf das Konfigurationsverzeichnis von Augustus schreiben, das solche Dateien enthält ( Augustus/config/ ). Wenn Sie Augustus weltweit in Ihrem System installieren, kann der config von allen Benutzern normalerweise nicht beschreibbar sein. Erstellen Sie entweder das Verzeichnis, in dem sich config rekursiv an die Benutzer von Augustus beschreibt, oder kopieren Sie den config/ den Ordner (rekursiv) an einen Ort, an dem Benutzer die Berechtigung schreiben.
Augustus sucht den config , indem er nach einer Umgebungsvariablen $AUGUSTUS_CONFIG_PATH sucht. Wenn die Umgebungsvariable von $AUGUSTUS_CONFIG_PATH nicht festgelegt ist, wird der Braker in den Weg nach dem ../config , in dem es eine ausführbare Augustus findet. Alternativ können Sie die Variable als Befehlszeilenargument an Braker angeben ( --AUGUSTUS_CONFIG_PATH=/your_path_to_AUGUSTUS/Augustus/config/ ). Wir empfehlen Ihnen, die variable EG für Ihre aktuelle Bash -Sitzung zu exportieren:
export AUGUSTUS_CONFIG_PATH=/your_path_to_AUGUSTUS/Augustus/config/
Um die Variable für alle Bash -Sitzungen zur Verfügung zu stellen, fügen Sie die obige Zeile einem Startskript hinzu, z. B. ~/.bashrc .
Bitte sehen Sie sich die Dockerfile an, falls Sie Augustus als Debian -Paket installieren möchten. Eine Reihe von Skripten muss dann gepatcht werden.
Braker erwartet das gesamte config von Augustus bei $AUGUSTUS_CONFIG_PATH , dh den species mit ihren Inhalten (zumindest generic ) und extrinsic ! Die Bereitstellung eines beschreibbaren, aber leeren Ordners bei $AUGUSTUS_CONFIG_PATH funktioniert für Braker nicht. Wenn Sie Augustus Binary und $AUGUSTUS_CONFIG_PATH trennen müssen, empfehlen wir Ihnen, die nicht geschriebenen Konfigurationsinhalte rekursiv an einen beschreibbaren Ort zu kopieren.
Wenn Sie eine systemweite Installation von Augustus AT /usr/bin/augustus haben, befindet sich eine unkohelbare Kopie der config unter /usr/bin/augustus_config/ . Der Ordner /home/yours/ ist für Sie beschreibbar. Kopieren Sie mit dem folgenden Befehl (und setzen Sie zusätzlich die dann erforderlichen Variablen fest):
cp -r /usr/bin/Augustus/config/ /home/yours/
export AUGUSTUS_CONFIG_PATH=/home/yours/augustus_config
export AUGUSTUS_BIN_PATH=/usr/bin
export AUGUSTUS_SCRIPTS_PATH=/usr/bin/augustus_scripts
Durch das Hinzufügen von Augustus -Binärdateien und -Skripts zu Ihrer $PATH -Variablen können Ihr System automatisch diese Tools finden. Es ist keine Voraussetzung dafür, dass Braker dies ausführt, da Braker sie vom Ort einer anderen Umgebungsvariablen ( $AUGUSTUS_CONFIG_PATH ) erraten wird, oder beide Verzeichnisse können als Befehlszeilenargumente an braker.pl geliefert werden, aber wir empfehlen es. Fügen Sie sie zu Ihrer $PATH -Variablen hinzu. Geben Sie für Ihre aktuelle Bash -Sitzung ein:
PATH=:/your_path_to_augustus/bin/:/your_path_to_augustus/scripts/:$PATH
export PATH
Fügen Sie für alle Ihre Bash -Sitzungen die obigen Zeilen einem Startskript (z. B. ~/.bashrc ) hinzu.
Bei Ubuntu ist Python3 normalerweise standardmäßig installiert, python3 befindet sich standardmäßig in Ihrer $PATH -Variablen, und der Braker befindet sich automatisch. Sie haben jedoch die Möglichkeit, den Binärstandort python3 auf zwei andere Arten anzugeben:
Exportieren Sie eine Umgebungsvariable $PYTHON3_PATH , z. B. in Ihrer ~/.bashrc -Datei:
export PYTHON3_PATH=/path/to/python3/
Geben Sie die Befehlszeilenoption --PYTHON3_PATH=/path/to/python3/ to braker.pl .
Laden Sie BAMTOOLS herunter (zB git clone https://github.com/pezmaster31/bamtools.git ). Installieren Sie BAMTOOLS, indem Sie Folgendes in Ihre Shell eingeben:
cd your-bamtools-directory mkdir build cd build cmake .. make
Wenn der Braker bereits in Ihrer $PATH -Variablen ist, findet er automatisch Bamtools. Andernfalls kann Braker die Bamtools Binary entweder mit einer Umgebungsvariablen $BAMTOOLS_PATH oder durch die Übernahme eines Befehlszeilenarguments ( --BAMTOOLS_PATH=/your_path_to_bamtools/bin/ f6 ) finden. Um die Umgebungsvariable für Ihre aktuelle Bash -Sitzung festzulegen, geben Sie ein:
export BAMTOOLS_PATH=/your_path_to_bamtools/bin/
Fügen Sie die obige Zeile einem Startskript (z. B. ~/.bashrc ) hinzu, um die Umgebungsvariable für alle Bash -Sitzungen festzulegen.
Sie können entweder NCBI Blast+ oder Diamond verwenden, um redundante Trainingsgene zu entfernen. Sie benötigen nicht beide Werkzeuge. Wenn Diamond vorhanden ist, wird es bevorzugt, weil es viel schneller ist.
Erhalten und auspacken Diamant wie folgt:
wget http://github.com/bbuchfink/diamond/releases/download/v0.9.24/diamond-linux64.tar.gz
tar xzf diamond-linux64.tar.gz
Wenn der Braker bereits in Ihrer $PATH -Variablen ist, findet der Braker Diamond automatisch. Andernfalls kann Braker die Diamant -Binärin entweder mit einer Umgebungsvariablen $DIAMOND_PATH oder anhand eines Befehlszeilenarguments ( --DIAMOND_PATH=/your_path_to_diamond ) lokalisieren. Um die Umgebungsvariable für Ihre aktuelle Bash -Sitzung festzulegen, geben Sie ein:
export DIAMOND_PATH=/your_path_to_diamond/
Fügen Sie die obige Zeile einem Startskript (z. B. ~/.bashrc ) hinzu, um die Umgebungsvariable für alle Bash -Sitzungen festzulegen.
Wenn Sie sich für Blast+ entscheiden, installieren Sie NCBI BLAST+ mit sudo apt-get install ncbi-blast+ .
Wenn der Braker bereits in Ihrer $PATH -Variablen ist, findet Brawer BLASTP automatisch. Andernfalls kann Braker die BLASTP -Binärdie entweder mit einer Umgebungsvariablen $BLAST_PATH oder durch die Übernahme eines Befehlszeilenarguments ( --BLAST_PATH=/your_path_to_blast/ ) finden. Um die Umgebungsvariable für Ihre aktuelle Bash -Sitzung festzulegen, geben Sie ein:
export BLAST_PATH=/your_path_to_blast/
Fügen Sie die obige Zeile einem Startskript (z. B. ~/.bashrc ) hinzu, um die Umgebungsvariable für alle Bash -Sitzungen festzulegen.
Die folgenden Tools sind von Genemark-ETP erforderlich und versucht, sie in Ihrer $PATH Variablen zu finden. Stellen Sie also sicher, dass Sie ihren Standort zu Ihrem $PATH , z. B. hinzufügen:
export PATH=$PATH:/your/path/to/Tool
Fügen Sie für alle folgenden Tools die obige Zeile einem Startskript (z. B. ~/.bashrc ) hinzu, um Ihre $PATH -Variable für alle Bash -Sitzungen zu erweitern.
Diese Softwaretools sind nur obligatorisch, wenn Sie Braker mit RNA-Seq- und Proteindaten betreiben!
StringTie2 wird von Genemark-ETP zum Zusammenbau ausgerichteter RNA-Seq-Alignments verwendet. Eine vorkompilierte Version von StringTie2 kann von https://ccb.jhu.edu/software/stringtie/#install heruntergeladen werden.
Das Softwarepaket Bedtools ist von Genemark-ETP erforderlich, wenn Sie Braker sowohl mit RNA-Seq- als auch mit Proteindaten ausführen möchten. Sie können Bedtools von https://github.com/arq5x/bedtools2/releases herunterladen. Hier können Sie entweder eine vorkompilierte Version bedtools.static.binary , z.
wget https://github.com/arq5x/bedtools2/releases/download/v2.30.0/bedtools.static.binary
mv bedtools.static.binary bedtools
chmod a+x
oder Sie können bedtools-2.30.0.tar.gz herunterladen und sie von der Quelle make , z.
wget https://github.com/arq5x/bedtools2/releases/download/v2.30.0/bedtools-2.30.0.tar.gz
tar -zxvf bedtools-2.30.0.tar.gz
cd bedtools2
make
Weitere Informationen finden Sie unter https://bedtools.readthedocs.io/en/latest/content/installation.html.
GFFREAD ist eine von Genemark-ETP benötigte Versorgungssoftware. Es kann von https://github.com/gpertea/gffread/releases/download/v0.12.7/gffread-0.12.7.Linux_x86_64.tar.gz heruntergeladen und mit make installiert, z.
wget https://github.com/gpertea/gffread/releases/download/v0.12.7/gffread-0.12.7.Linux_x86_64.tar.gz
tar xzf gffread-0.12.7.Linux_x86_64.tar.gz
cd gffread-0.12.7.Linux_x86_64
make
Sampools ist nicht erforderlich, um Braker ohne Genemark-ETP zu leiten, wenn alle Ihre Dateien korrekt formatiert sind (dh alle Sequenzen sollten kurze und eindeutige Fasta-Namen haben). Wenn Sie sich nicht sicher sind, ob alle Ihre Dateien korrekt vorgenommen werden, ist es möglicherweise hilfreich, dass Samtools installiert werden, da Braker bestimmte Formatprobleme automatisch mit Samtools beheben kann.
Als Voraussetzung htslib SAMTOOLS laden Sie htslib herunter ( git clone https://github.com/samtools/htslib.git .
Download und installieren Sie SAMTOOLS (z. B. git clone git://github.com/samtools/samtools.git ) und folgen Sie anschließend die Dokumentation der SAMTOOLS für die Installation).
Wenn Braker bereits in Ihrer $PATH -Variable findet, findet er automatisch Samtools. Andernfalls kann Braker Samtools entweder durch ein Befehlszeilenargument ( --SAMTOOLS_PATH=/your_path_to_samtools/ ) oder mithilfe einer Umgebungsvariablen $SAMTOOLS_PATH finden. Zum Exportieren der Variablen, z. B. für Ihre aktuelle Bash -Sitzung, geben Sie ein:
export SAMTOOLS_PATH=/your_path_to_samtools/
Fügen Sie die obige Zeile einem Startskript (z. B. ~/.bashrc ) hinzu, um die Umgebungsvariable für alle Bash -Sitzungen festzulegen.
Wenn Biopython installiert ist, kann Braker Fasta-Files mit Codierungssequenzen und Proteinsequenzen erzeugen, die von Augustus vorhergesagt und Spurdaten-Hubs zur Visualisierung eines Braker-Laufs mit Makehub R16 erzeugen. Dies sind optionale Schritte. Das erste kann mit dem Befehlszeilen-Flag --skipGetAnnoFromFasta deaktiviert werden. Der zweite kann mithilfe der Befehlszeilenoptionen --makehub [email protected] , Biopython nicht erforderlich ist, aktiviert werden, wenn keiner dieser optionalen Schritte erforderlich ist soll durchgeführt werden.
Installieren Sie bei Ubuntu Python3 Paket Manager mit:
`sudo apt-get install python3-pip`
Installieren Sie dann Biopython mit:
`sudo pip3 install biopython`
Cdbfasta und CDByank werden von Braker benötigt, um Augustus -Gene mit in Frame -Stop -Codons (Spleiß -Stopp -Codons) mit dem Augustus -Skript FIX_IN_FRAME_STOP_CODON_GENES.PY zu korrigieren. Dies kann mit --skip_fixing_broken_genes übersprungen werden.
Installieren Sie auf Ubuntu CDBasta mit:
sudo apt-get install cdbfasta
Für andere Systeme können Sie beispielsweise CDBfasta von https://github.com/gpertea/cdbfasta, z. B. erhalten:
git clone https://github.com/gpertea/cdbfasta.git
cd cdbfasta
make all
Auf Ubuntu befinden sich CDBasta und CDByank nach der Installation in Ihrer $PATH -Variablen, und der Braker findet sie automatisch. Sie haben jedoch die Möglichkeit, den Binärstandort cdbfasta und cdbyank auf zwei andere Arten anzugeben:
$CDBTOOLS_PATH , z. B. in Ihrer ~/.bashrc -Datei: export CDBTOOLS_PATH=/path/to/cdbtools/
--CDBTOOLS_PATH=/path/to/cdbtools/ TO braker.pl . Hinweis: Die Unterstützung des eigenständigen Spaln (OUside von Prothint) innerhalb des Brakers ist veraltet.
Dieses Tool ist erforderlich, wenn Sie Prothint ausführen oder wenn Sie Protein zu Genomausrichtungen mit Braker mit Spaln außerhalb von Prothint ausführen möchten. Die Verwendung von Spaln außerhalb von Prothint ist nur dann ein geeigneter Ansatz, wenn eine kommentierte Art von kurzer evolutionärer Entfernung zu Ihrem Zielgenom verfügbar ist. Wir empfehlen, Spaln durch Prothint für Braker zu führen. Prothint bringt eine Spaln -Binärin mit. Wenn das nicht auf Ihrem System funktioniert, laden Sie Spaln von https://github.com/ogotoh/spaln herunter. Entpacken und installieren Sie gemäß spaln/doc/SpalnReadMe22.pdf .
Braker versucht, die ausführbare Spaln -Datei mithilfe einer Umgebungsvariablen $ALIGNMENT_TOOL_PATH zu finden. Alternativ kann dies als Befehlszeilenargument ( --ALIGNMENT_TOOL_PATH=/your/path/to/spaln ) geliefert werden.
Dieses Tool ist nur erforderlich, wenn Sie entweder UTRs (RNA-Seq-Daten) zu prognostizierten Genen hinzufügen möchten oder wenn Sie UTR-Parameter für Augustus ausbilden und Gene mit UTRs vorhergesagt haben. In jedem Fall benötigt Gushr die Eingabe von RNA-Seq-Daten.
Gushr kann unter https://github.com/gaius-augustus/gushr heruntergeladen werden. Erhalten Sie es durch Eingabe:
git clone https://github.com/Gaius-Augustus/GUSHR.git
Gushr führt eine Gemoma JAR -Datei R19, R20, R21 aus, und diese JAR -Datei erfordert Java 1.8. Auf Ubuntu können Sie Java 1.8 mit dem folgenden Befehl installieren:
sudo apt-get install openjdk-8-jdk
Wenn Sie mehrere Java -Versionen auf Ihrem System installiert haben, sollten Sie sicherstellen
sudo update-alternatives --config java
und Auswahl der richtigen Version.
Wenn Sie wechseln --UTR=on , benötigt BAMTOWIG.PY die folgenden Tools, die von http://hgdownload.soe.ucsc.edu/admin/exe heruntergeladen werden können:
TWOBITINFO
FatotWobit
Es ist optional, diese Tools in Ihrem $ -Path zu installieren. Wenn Sie dies nicht tun und Sie wechseln --UTR=on , wird BAMTOWIG.PY sie automatisch in das Arbeitsverzeichnis herunterladen.
Wenn Sie automatisch einen Track-Datenzentrum Ihres Braker-Laufs generieren möchten, ist die Makehub-Software, die unter https://github.com/gaius-augustus/makehub verfügbar ist, erforderlich. Laden Sie die Software herunter (entweder durch Ausführen git clone https://github.com/Gaius-Augustus/MakeHub.git oder durch Auswahl einer Version von https://github.com/gaius-augustus/makehub/releases. Paket, wenn Sie eine Version heruntergeladen haben (z. B. unzip MakeHub.zip oder tar -zxvf MakeHub.tar.gz .
Braker versucht, das Skript make_hub.py mit einer Umgebungsvariablen $MAKEHUB_PATH zu finden. Alternativ kann dies als Befehlszeilenargument geliefert werden ( --MAKEHUB_PATH=/your/path/to/MakeHub/ ). Braker kann auch versuchen, den Standort von Makehub in Ihrem System zu erraten.
Wenn Sie möchten, dass Braker RNA-Seq-Bibliotheken von der SRA von NCBI herunterlädt, ist das SRA-Toolkit erforderlich. Sie können eine vorkompilierte Version des SRA-Toolkits von http://daehwankimlab.github.io/hisat2/download/version-hisat2-221 erhalten.
Braker versucht, ausführbare Binärdateien aus dem SRA-Toolkit (Fastq-Dump, Prefetch) zu finden, indem eine Umgebungsvariable $SRATOOLS_PATH verwendet wird. Alternativ kann dies als Befehlszeilenargument geliefert werden ( --SRATOOLS_PATH=/your/path/to/SRAToolkit/ ). Braker kann auch versuchen, den Standort des SRA -Toolkits auf Ihrem System zu erraten, wenn sich die ausführbaren Variablen in Ihrer $PATH -Variablen befinden.
Wenn Sie nicht ausgerichtete RNA-Seq-Lesevorgänge verwenden möchten, muss die HISAT2-Software dem Genom zugeordnet werden. Eine vorkompilierte Version von HISAT2 kann von http://daehwankimlab.github.io/hisat2/download/version-hisat2-221 heruntergeladen werden.
Braker wird versuchen, ausführbare HISAT2-Binärdateien (Hisat2, Hisat2-Build) zu finden, indem eine Umgebungsvariable $HISAT2_PATH verwendet wird. Alternativ kann dies als Befehlszeilenargument geliefert werden ( --HISAT2_PATH=/your/path/to/HISAT2/ ). Braker kann auch versuchen, den Standort von Hisat2 in Ihrem System zu erraten, wenn sich die ausführbaren Variablen in Ihrer $PATH -Variablen befinden.
Wenn Sie Tsebra im Braker in einem Maximierungsmodus für Busco -Vollständigkeit ausführen möchten, müssen Sie Complasm installieren.
wget https://github.com/huangnengCSU/compleasm/releases/download/v0.2.4/compleasm-0.2.4_x64-linux.tar.bz2
tar -xvjf compleasm-0.2.4_x64-linux.tar.bz2 &&
Fügen Sie die resultierende Ordner conpeasm_kit zu Ihrer $PATH -Variablen, z. B. hinzu:
export PATH=$PATH:/your/path/to/compleasm_kit
CHORPASM erfordert Pandas, die mit:
pip install pandas
Braker (Braker.pl) verwendet GetConf, um zu sehen, wie viele Threads auf Ihrem System ausgeführt werden können. Auf Ubuntu können Sie es installieren mit:
sudo apt-get install libc-bin
Im Folgenden beschreiben wir „typische“ Braker -Forderungen für unterschiedliche Eingabedatentypen. Im Allgemeinen empfehlen wir Ihnen, Braker über genomische Sequenzen zu betreiben, die für Wiederholungen Softmasked wurden. Braker sollte nur auf Genome angewendet werden, die für Wiederholungen weichmaskiert wurden!
This approach is suitable for genomes of species for which RNA-Seq libraries with good transcriptome coverage are available and for which protein data is not at hand. The pipeline is illustrated in Figure 2.
BRAKER has several ways to receive RNA-Seq data as input:
You can provide ID(s) of RNA-Seq libraries from SRA (in case of multiple IDs, separate them by comma) as argument to --rnaseq_sets_ids . The libraries belonging to the IDs are then downloaded automatically by BRAKER, eg:
braker.pl --species=yourSpecies --genome=genome.fasta
--rnaseq_sets_ids=SRA_ID1,SRA_ID2
You can use local FASTQ file(s) of unaligned reads as input. In this case, you have to provide BRAKER with the ID(s) of the RNA-Seq set(s) as argument to --rnaseq_sets_ids and the path(s) to the directories, where the FASTQ files are located as argument to --rnaseq_sets_dirs . For each ID ID , BRAKER will search in these directories for one FASTQ file named ID.fastq if the reads are unpaired, or for two FASTQ files named ID_1.fastq and ID_2.fastq if they are paired.
For example, if you have a paired library called 'SRA_ID1' and an unpaired library named 'SRA_ID2', you have to have a directory /path/to/local/fastq/files/ , where the files SRA_ID1_1.fastq , SRA_ID1_2.fastq , and SRA_ID2.fastq reside. Then, you could run BRAKER with following command:
braker.pl --species=yourSpecies --genome=genome.fasta
--rnaseq_sets_ids=SRA_ID1,SRA_ID2
--rnaseq_sets_dirs=/path/to/local/fastq/files/
There are two ways of supplying BRAKER with RNA-Seq data as bam file(s). First, you can do it in the same way as you would supply FASTQ file(s): Provide the ID(s)/name(s) of your bam file(s) as argument to --rnaseq_sets_ids and specify directories where the bam files reside with --rnaseq_sets_dirs . BRAKER will automatically detect that these ID(s) are bam and not FASTQ file(s), eg:
braker.pl --species=yourSpecies --genome=genome.fasta
--rnaseq_sets_ids=BAM_ID1,BAM_ID2
--rnaseq_sets_dirs=/path/to/local/bam/files/
Second, you can specify the paths to your bam file(s) directly, eg can either extract RNA-Seq spliced alignment information from bam files, or it can use such extracted information, directly.
braker.pl --species=yourSpecies --genome=genome.fasta
--bam=file1.bam,file2.bam
Please note that we generally assume that bam files were generated with HiSat2 because that is the aligner that would also be executed by BRAKER3 with fastq input. If you want for some reason to generate the bam files with STAR, use the option --outSAMstrandField intronMotif of STAR to produce files that are compatible wiht StringTie in BRAKER3.
In order to run BRAKER with RNA-Seq spliced alignment information that has already been extracted, run:
braker.pl --species=yourSpecies --genome=genome.fasta
--hints=hints1.gff,hints2.gff
The format of such a hints file must be as follows (tabulator separated file):
chrName b2h intron 6591 8003 1 + . pri=4;src=E
chrName b2h intron 6136 9084 11 + . mult=11;pri=4;src=E
...
The source b2h in the second column and the source tag src=E in the last column are essential for BRAKER to determine whether a hint has been generated from RNA-Seq data.
It is also possible to provide RNA-Seq sets in different ways for the same BRAKER run, any combination of above options is possible. It is not recommended to provide RNA-Seq data with --hints if you run BRAKER in ETPmode (RNA-Seq and protein data), because GeneMark-ETP won't use these hints!
This approach is suitable for genomes of species for which no RNA-Seq libraries are available. A large database of proteins (with possibly longer evolutionary distance to the target species) should be used in this case. This mode is illustrated in figure 9.
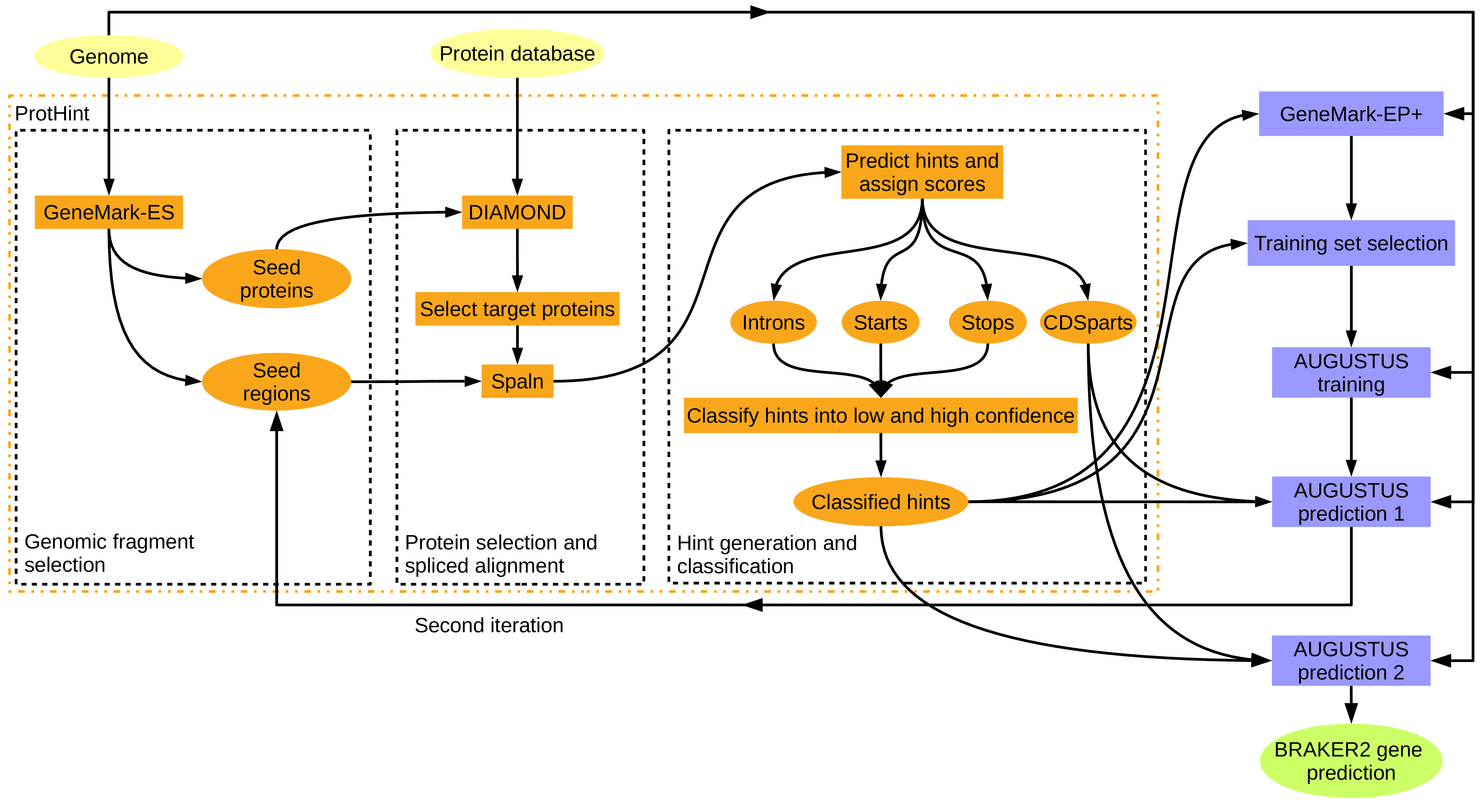
Figure 9: BRAKER with proteins of any evolutionary distance. ProtHint protein mapping pipelines is used to generate protein hints. ProtHint automatically determines which alignments are from close relatives, and which are from rather distant relatives.
For running BRAKER in this mode, type:
braker.pl --genome=genome.fa --prot_seq=proteins.fa
We recommend using OrthoDB as basis for proteins.fa . The instructions on how to prepare the input OrthoDB proteins are documented here: https://github.com/gatech-genemark/ProtHint#protein-database-preparation.
You can of course add additional protein sequences to that file, or try with a completely different database. Any database will need several representatives for each protein, though.
Instead of having BRAKER run ProtHint, you can also start BRAKER with hints already produced by ProtHint, by providing ProtHint's prothint_augustus.gff output:
braker.pl --genome=genome.fa --hints=prothint_augustus.gff
The format of prothint_augustus.gff in this mode looks like this:
2R ProtHint intron 11506230 11506648 4 + . src=M;mult=4;pri=4
2R ProtHint intron 9563406 9563473 1 + . grp=69004_0:001de1_702_g;src=C;pri=4;
2R ProtHint intron 8446312 8446371 1 + . grp=43151_0:001cae_473_g;src=C;pri=4;
2R ProtHint intron 8011796 8011865 2 - . src=P;mult=1;pri=4;al_score=0.12;
2R ProtHint start 234524 234526 1 + . src=P;mult=1;pri=4;al_score=0.08;
The prediction of all hints with src=M will be enforced. Hints with src=C are 'chained evidence', ie they will only be incorporated if all members of the group (grp=...) can be incorporated in a single transcript. All other hints have src=P in the last column. Supported features in column 3 are intron , start , stop and CDSpart .
If RNA-Seq (and only RNA-Seq) data is provided to BRAKER as a bam-file, and if the genome is softmasked for repeats, BRAKER can automatically train UTR parameters for AUGUSTUS. After successful training of UTR parameters, BRAKER will automatically predict genes including coverage information form RNA-Seq data. Example call:
braker.pl --species=yourSpecies --genome=genome.fasta
--bam=file.bam --UTR=on
Warnings:
This feature is experimental!
--UTR=on is currently not compatible with bamToWig.py as released in AUGUSTUS 3.3.3; it requires the current development code version from the github repository (git clone https://github.com/Gaius-Augustus/Augustus.git).
--UTR=on increases memory consumption of AUGUSTUS. Carefully monitor jobs if your machine was close to maxing RAM without --UTR=on! Reducing the number of cores will also reduce RAM consumption.
UTR prediction sometimes improves coding sequence prediction accuracy, but not always. If you try this feature, carefully compare results with and without UTR parameters, afterwards (eg in UCSC Genome Browser).
For running BRAKER without UTR parameters, it is not very important whether RNA-Seq data was generated by a stranded protocol (because spliced alignments are 'artificially stranded' by checking the splice site pattern). However, for UTR training and prediction, stranded libraries may provide information that is valuable for BRAKER.
After alignment of the stranded RNA-Seq libraries, separate the resulting bam file entries into two files: one for plus strand mappings, one for minus strand mappings. Call BRAKER as follows:
braker.pl --species=yourSpecies --genome=genome.fasta
--bam=plus.bam,minus.bam --stranded=+,-
--UTR=on
You may additionally include bam files from unstranded libraries. Those files will not used for generating UTR training examples, but they will be included in the final gene prediction step as unstranded coverage information, example call:
braker.pl --species=yourSpecies --genome=genome.fasta
--bam=plus.bam,minus.bam,unstranded.bam
--stranded=+,-,. --UTR=on
Warning: This feature is experimental and currently has low priority on our maintenance list!
The native mode for running BRAKER with RNA-Seq and protein data. This will call GeneMark-ETP, which will use RNA-Seq and protein hints for training GeneMark-ETP. Subsequently, AUGUSTUS is trained on 'high-confindent' genes (genes with very high extrinsic evidence support) from the GeneMark-ETP prediction and a set of genes is predicted by AUGUSTUS. In a last step, the predictions of AUGUSTUS and GeneMark-ETP are combined using TSEBRA.
Alignment of RNA-Seq reads
GeneMark-ETP utilizes Stringtie2 to assemble RNA-Seq data, which requires that the aligned reads (BAM files) contain the XS (strand) tag for spliced reads. Therefore, if you align your reads with HISAT2, you must enable the --dta option, or if you use STAR, you must use the --outSAMstrandField intronMotif option. TopHat alignments include this tag by default.
To call the pipeline in this mode, you have to provide it with a protein database using --prot_seq (as described in BRAKER with protein data), and RNA-Seq data either by their SRA ID so that they are downloaded by BRAKER, as unaligned reads in FASTQ format, and/or as aligned reads in bam format (as described in BRAKER with RNA-Seq data). You could also specify already processed extrinsic evidence using the --hints option. However, this is not recommend for a normal BRAKER run in ETPmode, as these hints won't be used in the GeneMark-ETP step. Only use --hints when you want to skip the GenMark-ETP step!
Examples of how you could run BRAKER in ETPmode:
braker.pl --genome=genome.fa --prot_seq=orthodb.fa
--rnaseq_sets_ids=SRA_ID1,SRA_ID2
--rnaseq_sets_dirs=/path/to/local/RNA-Seq/files/
braker.pl --genome=genome.fa --prot_seq=orthodb.fa
--rnaseq_sets_ids=SRA_ID1,SRA_ID2,SRA_ID3
braker.pl --genome=genome.fa --prot_seq=orthodb.fa
--bam=/path/to/SRA_ID1.bam,/path/to/SRA_ID2.bam
A preliminary protocol for integration of assembled subreads from PacBio ccs sequencing in combination with short read Illumina RNA-Seq and protein database is described at https://github.com/Gaius-Augustus/BRAKER/blob/master/docs/long_reads/long_read_protocol .md
We forked GeneMark-ETP and hard coded that StringTie will perform long read assembly in that particular version. If you want to use this 'fast-hack' version for BRAKER, you have to prepare the BAM file with long read to genome spliced alignments outside of BRAKER, eg:
T=48 # adapt to your number of threads
minimap2 -t${T} -ax splice:hq -uf genome.fa isoseq.fa > isoseq.sam
samtools view -bS --threads ${T} isoseq.sam -o isoseq.bam
Pull the adapted container:
singularity build braker3_lr.sif docker://teambraker/braker3:isoseq
Calling BRAKER3 with a BAM file of spliced-aligned IsoSeq Reads:
singularity exec -B ${PWD}:${PWD} braker3_lr.sif braker.pl --genome=genome.fa --prot_seq=protein_db.fa –-bam=isoseq.bam --threads=${T}
Warning Do NOT mix short read and long read data in this BRAKER/GeneMark-ETP variant!
Warning The accuracy of gene prediction here heavily depends on the depth of your isoseq data. We verified with PacBio HiFi reads from 2022 that given sufficient completeness of the assembled transcriptome you will reach similar results as with short reads. However, we also observed a drop in accuracy compared to short reads when using other long read data sets with higher error rates and less sequencing depth.
Please run braker.pl --help to obtain a full list of options.
Compute AUGUSTUS ab initio predictions in addition to AUGUSTUS predictions with hints (additional output files: augustus.ab_initio.* . This may be useful for estimating the quality of training gene parameters when inspecting predictions in a Browser.
One or several command line arguments to be passed to AUGUSTUS, if several arguments are given, separate them by whitespace, ie "--first_arg=sth --second_arg=sth" . This may be be useful if you know that gene prediction in your particular species benefits from a particular AUGUSTUS argument during the prediction step.
Specifies the maximum number of threads that can be used during computation. BRAKER has to run some steps on a single thread, others can take advantage of multiple threads. If you use more than 8 threads, this will not speed up all parallelized steps, in particular, the time consuming optimize_augustus.pl will not use more than 8 threads. However, if you don't mind some threads being idle, using more than 8 threads will speed up other steps.
GeneMark-ETP option: run algorithm with branch point model. Use this option if you genome is a fungus.
Use the present config and parameter files if they exist for 'species'; will overwrite original parameters if BRAKER performs an AUGUSTUS training.
Execute CRF training for AUGUSTUS; resulting parameters are only kept for final predictions if they show higher accuracy than HMM parameters. This increases runtime!
Change the parameter
Generate UTR training examples for AUGUSTUS from RNA-Seq coverage information, train AUGUSTUS UTR parameters and predict genes with AUGUSTUS and UTRs, including coverage information for RNA-Seq as evidence. This is an experimental feature!
If you performed a BRAKER run without --UTR=on, you can add UTR parameter training and gene prediction with UTR parameters (and only RNA-Seq hints) with the following command:
braker.pl --genome=../genome.fa --addUTR=on
--bam=../RNAseq.bam --workingdir=$wd
--AUGUSTUS_hints_preds=augustus.hints.gtf
--threads=8 --skipAllTraining --species=somespecies
Modify augustus.hints.gtf to point to the AUGUSTUS predictions with hints from previous BRAKER run; modify flaning_DNA value to the flanking region from the log file of your previous BRAKER run; modify some_new_working_directory to the location where BRAKER should store results of the additional BRAKER run; modify somespecies to the species name used in your previous BRAKER run.
Add UTRs from RNA-Seq converage information to AUGUSTUS gene predictions using GUSHR. No training of UTR parameters and no gene prediction with UTR parameters is performed.
If you performed a BRAKER run without --addUTR=on, you can add UTRs results of a previous BRAKER run with the following command:
braker.pl --genome=../genome.fa --addUTR=on
--bam=../RNAseq.bam --workingdir=$wd
--AUGUSTUS_hints_preds=augustus.hints.gtf --threads=8
--skipAllTraining --species=somespecies
Modify augustus.hints.gtf to point to the AUGUSTUS predictions with hints from previous BRAKER run; modify some_new_working_directory to the location where BRAKER should store results of the additional BRAKER run; this run will not modify AUGUSTUS parameters. We recommend that you specify the original species of the original run with --species=somespecies . Otherwise, BRAKER will create an unneeded species parameters directory Sp_* .
If --UTR=on is enabled, strand-separated bam-files can be provided with --bam=plus.bam,minus.bam . In that case, --stranded=... should hold the strands of the bam files ( + for plus strand, - for minus strand, . for unstranded). Note that unstranded data will be used in the gene prediction step, only, if the parameter --stranded=... is set. This is an experimental feature! GUSHR currently does not take advantage of stranded data.
If --makehub and [email protected] (with your valid e-mail adress) are provided, a track data hub for visualizing results with the UCSC Genome Browser will be generated using MakeHub (https://github.com/Gaius-Augustus/MakeHub).
By default, GeneMark-ES/ET/EP/ETP uses a probability of 0.001 for predicting the donor splice site pattern GC (instead of GT). It may make sense to increase this value for species where this donor splice site is more common. For example, in the species Emiliania huxleyi , about 50% of donor splice sites have the pattern GC (https://media.nature.com/original/nature-assets/nature/journal/v499/n7457/extref/nature12221-s2.pdf, page 5).
Use a species-specific lineage, eg arthropoda_odb10 for an arthropod. BRAKER does not support auto-typing of the lineage.
Specifying a BUSCO-lineage invokes two changes in BRAKER R28 :
BRAKER will run compleasm with the specified lineage in genome mode and convert the detected BUSCO matches into hints for AUGUSTUS. This may increase the number of BUSCOs in the augustus.hints.gtf file slightly.
BRAKER will invoke best_by_compleasm.py to check whether the braker.gtf file that is by default generated by TSEBRA has the lowest amount of missing BUSCOs compared to the augustus.hints.gtf and the genemark.gtf file. If not, the following decision schema is applied to re-run TSEBRA to minimize the missing BUSCOs in the final output of BRAKER (always braker.gtf). If an alternative and better gene set is created, the original braker.gtf gene set is moved to a directory called braker_original. Information on what happened during the best_by_compleasm.py run is written to the file best_by_compleasm.log.
![best_by_busco[fig14]](https://images.downcodes.com/uploads/20250214/img_67aee79a11fd439.png)
Please note that using BUSCO to assess the quality of a gene set, in particular when comparing BRAKER to other pipelines, does not make sense once you specified a BUSCO lineage. We recommend that you use other measures to assess the quality of your gene set, eg by comparing it to a reference gene set or running OMArk.
BRAKER produces several important output files in the working directory.
braker.gtf: Final gene set of BRAKER. This file may contain different contents depending on how you called BRAKER
in ETPmode: Final gene set of BRAKER consisting of genes predicted by AUGUSTUS and GeneMark-ETP that were combined and filtered by TSEBRA.
otherwise: Union of augustus.hints.gtf and reliable GeneMark-ES/ET/EP predictions (genes fully supported by external evidence). In --esmode , this is the union of augustus.ab_initio.gtf and all GeneMark-ES genes. Thus, this set is generally more sensitive (more genes correctly predicted) and can be less specific (more false-positive predictions can be present). This output is not necessarily better than augustus.hints.gtf, and it is not recommended to use it if BRAKER was run in ESmode.
braker.codingseq: Final gene set with coding sequences in FASTA format
braker.aa: Final gene set with protein sequences in FASTA format
braker.gff3: Final gene set in gff3 format (only produced if the flag --gff3 was specified to BRAKER.
Augustus/*: Augustus gene set(s) in as gtf/conding/aa files
GeneMark-E*/genemark.gtf: Genes predicted by GeneMark-ES/ET/EP/EP+/ETP in GTF-format.
hintsfile.gff: The extrinsic evidence data extracted from RNAseq.bam and/or protein data.
braker_original/*: Genes predicted by BRAKER (TSEBRA merge) before compleasm was used to improve BUSCO completeness
bbc/*: output folder of best_by_compleasm.py script from TSEBRA that is used to improve BUSCO completeness in the final output of BRAKER
Output files may be present with the following name endings and formats:
Coding sequences in FASTA-format are produced if the flag --skipGetAnnoFromFasta was not set.
Protein sequence files in FASTA-format are produced if the flag --skipGetAnnoFromFasta was not set.
For details about gtf format, see http://www.sanger.ac.uk/Software/formats/GFF/. A GTF-format file contains one line per predicted exon. Beispiel:
HS04636 AUGUSTUS initial 966 1017 . + 0 transcript_id "g1.1"; gene_id "g1";
HS04636 AUGUSTUS internal 1818 1934 . + 2 transcript_id "g1.1"; gene_id "g1";
The columns (fields) contain:
seqname source feature start end score strand frame transcript ID and gene ID
If the --makehub option was used and MakeHub is available on your system, a hub directory beginning with the name hub_ will be created. Copy this directory to a publicly accessible web server. A file hub.txt resides in the directory. Provide the link to that file to the UCSC Genome Browser for visualizing results.
An incomplete example data set is contained in the directory BRAKER/example . In order to complete the data set, please download the RNA-Seq alignment file (134 MB) with wget :
cd BRAKER/example
wget http://topaz.gatech.edu/GeneMark/Braker/RNAseq.bam
In case you have trouble accessing that file, there's also a copy available from another server:
cd BRAKER/example
wget http://bioinf.uni-greifswald.de/augustus/datasets/RNAseq.bam
The example data set was not compiled in order to achieve optimal prediction accuracy, but in order to quickly test pipeline components. The small subset of the genome used in these test examples is not long enough for BRAKER training to work well.
Data corresponds to the last 1,000,000 nucleotides of Arabidopsis thaliana 's chromosome Chr5, split into 8 artificial contigs.
RNA-Seq alignments were obtained by VARUS.
The protein sequences are a subset of OrthoDB v10 plants proteins.
List of files:
genome.fa - genome file in fasta formatRNAseq.bam - RNA-Seq alignment file in bam format (this file is not a part of this repository, it must be downloaded separately from http://topaz.gatech.edu/GeneMark/Braker/RNAseq.bam)RNAseq.hints - RNA-Seq hints (can be used instead of RNAseq.bam as RNA-Seq input to BRAKER)proteins.fa - protein sequences in fasta formatThe below given commands assume that you configured all paths to tools by exporting bash variables or that you have the necessary tools in your $PATH.
The example data set also contains scripts tests/test*.sh that will execute below listed commands for testing BRAKER with the example data set. You find example results of AUGUSTUS and GeneMark-ES/ET/EP/ETP in the folder results/test* . Be aware that BRAKER contains several parts where random variables are used, ie results that you obtain when running the tests may not be exactly identical. To compare your test results with the reference ones, you can use the compare_intervals_exact.pl script as follows:
# Compare CDS features
compare_intervals_exact.pl --f1 augustus.hints.gtf --f2 ../../results/test${N}/augustus.hints.gtf --verbose
# Compare transcripts
compare_intervals_exact.pl --f1 augustus.hints.gtf --f2 ../../results/test${N}/augustus.hints.gtf --trans --verbose
Several tests use --gm_max_intergenic 10000 option to make the test runs faster. It is not recommended to use this option in real BRAKER runs, the speed increase achieved by adjusting this option is negligible on full-sized genomes.
We give runtime estimations derived from computing on Intel(R) Xeon(R) CPU E5530 @ 2.40GHz .
The following command will run the pipeline according to Figure 3:
braker.pl --genome genome.fa --bam RNAseq.bam --threads N --busco_lineage=lineage_odb10
This test is implemented in test1.sh , expected runtime is ~20 minutes.
The following command will run the pipeline according to Figure 4:
braker.pl --genome genome.fa --prot_seq proteins.fa --threads N --busco_lineage=lineage_odb10
This test is implemented in test2.sh , expected runtime is ~20 minutes.
The following command will run a pipeline that first trains GeneMark-ETP with protein and RNA-Seq hints and subsequently trains AUGUSTUS on the basis of GeneMark-ETP predictions. AUGUSTUS predictions are also performed with hints from both sources, see Figure 5.
Run with local RNA-Seq file:
braker.pl --genome genome.fa --prot_seq proteins.fa --bam ../RNAseq.bam --threads N --busco_lineage=lineage_odb10
This test is implemented in test3.sh , expected runtime is ~20 minutes.
Download RNA-Seq library from Sequence Read Archive (~1gb):
braker.pl --genome genome.fa --prot_seq proteins.fa --rnaseq_sets_ids ERR5767212 --threads N --busco_lineage=lineage_odb10
This test is implemented in test3_4.sh , expected runtime is ~35 minutes.
The training step of all pipelines can be skipped with the option --skipAllTraining . This means, only AUGUSTUS predictions will be performed, using pre-trained, already existing parameters. For example, you can predict genes with the command:
braker.pl --genome=genome.fa --bam RNAseq.bam --species=arabidopsis
--skipAllTraining --threads N
This test is implemented in test4.sh , expected runtime is ~1 minute.
The following command will run the pipeline with no extrinsic evidence:
braker.pl --genome=genome.fa --esmode --threads N
This test is implemented in test5.sh , expected runtime is ~20 minutes.
The following command will run BRAKER with training UTR parameters from RNA-Seq coverage data:
braker.pl --genome genome.fa --bam RNAseq.bam --UTR=on --threads N
This test is implemented in test6.sh , expected runtime is ~20 minutes.
The following command will add UTRs to augustus.hints.gtf from RNA-Seq coverage data:
braker.pl --genome genome.fa --bam RNAseq.bam --addUTR=on --threads N
This test is implemented in test7.sh , expected runtime is ~20 minutes.
There is currently no clean way to restart a failed BRAKER run (after solving some problem). However, it is possible to start a new BRAKER run based on results from a previous run -- given that the old run produced the required intermediate results. We will in the following refer to the old working directory with variable ${BRAKER_OLD} , and to the new BRAKER working directory with ${BRAKER_NEW} . The file what-to-cite.txt will always only refer to the software that was actually called by a particular run. You might have to combine the contents of ${BRAKER_NEW}/what-to-cite.txt with ${BRAKER_OLD}/what-to-cite.txt for preparing a publication. The following figure illustrates at which points BRAKER run may be intercepted.
![braker-intercept[fig8]](https://images.downcodes.com/uploads/20250214/img_67aee79a12cab310.png)
Figure 10: Points for intercepting a BRAKER run and reusing intermediate results in a new BRAKER run.
This option is only possible for BRAKER in ETmode or EPmode and nicht in ETPmode!
If you have access to an existing BRAKER output that contains hintsfiles that were generated from extrinsic data, such as RNA-Seq or protein sequences, you can recycle these hints files in a new BRAKER run. Also, hints from a separate ProtHint run can be directly used in BRAKER.
The hints can be given to BRAKER with --hints ${BRAKER_OLD}/hintsfile.gff option. This is illustrated in the test files test1_restart1.sh , test2_restart1.sh , test4_restart1.sh . The other modes (for which this test is missing) cannot be restarted in this way.
The GeneMark result can be given to BRAKER with --geneMarkGtf ${BRAKER_OLD}/GeneMark*/genemark.gtf option if BRAKER is run in ETmode or EPmode. This is illustrated in the test files test1_restart2.sh , test2_restart2.sh , test5_restart2.sh .
In ETPmode, you can either provide BRAKER with the results of the GeneMarkETP step manually, with --geneMarkGtf ${BRAKER_OLD}/GeneMark-ETP/proteins.fa/genemark.gtf , --traingenes ${BRAKER_OLD}/GeneMark-ETP/training.gtf , and --hints ${BRAKER_OLD}/hintsfile.gff (see test3_restart1.sh for an example), or you can specify the previous GeneMark-ETP results with the option --gmetp_results_dir ${BRAKER_OLD}/GeneMark-ETP/ so that BRAKER can search for the files automatically (see test3_restart2.sh for an example).
The trained species parameters for AGUSTUS can be passed with --skipAllTraining and --species $speciesName options. This is illustrated in test*_restart3.sh files. Note that in ETPmode you have to specify the GeneMark files as described in Option 2!
Before reporting bugs, please check that you are using the most recent versions of GeneMark-ES/ET/EP/ETP, AUGUSTUS and BRAKER. Also, check the list of Common problems, and the Issue list on GitHub before reporting bugs. We do monitor open issues on GitHub. Sometimes, we are unable to help you, immediately, but we try hard to solve your problems.
If you found a bug, please open an issue at https://github.com/Gaius-Augustus/BRAKER/issues (or contact [email protected] or [email protected]).
Information worth mentioning in your bug report:
Check in braker/yourSpecies/braker.log at which step braker.pl crashed.
There are a number of other files that might be of interest, depending on where in the pipeline the problem occurred. Some of the following files will not be present if they did not contain any errors.
braker/yourSpecies/errors/bam2hints.*.stderr - will give details on a bam2hints crash (step for converting bam file to intron gff file)
braker/yourSpecies/hintsfile.gff - is this file empty? If yes, something went wrong during hints generation - does this file contain hints from source “b2h” and of type “intron”? If not: GeneMark-ET will not be able to execute properly. Conversely, GeneMark-EP+ will not be able to execute correctly if hints from the source "ProtHint" are missing.
braker/yourSpecies/spaln/*err - errors reported by spaln
braker/yourSpecies/errors/GeneMark-{ET,EP,ETP}.stderr - errors reported by GeneMark-ET/EP+/ETP
braker/yourSpecies/errors/GeneMark-{ET,EP,ETP).stdout - may give clues about the point at which errors in GeneMark-ET/EP+/ETP occured
braker/yourSpecies/GeneMark-{ET,EP,ETP}/genemark.gtf - is this file empty? If yes, something went wrong during executing GeneMark-ET/EP+/ETP
braker/yourSpecies/GeneMark-{ET,EP}/genemark.f.good.gtf - is this file empty? If yes, something went wrong during filtering GeneMark-ET/EP+ genes for training AUGUSTUS
braker/yourSpecies/genbank.good.gb - try a “grep -c LOCUS genbank.good.gb” to determine the number of training genes for training AUGUSTUS, should not be low
braker/yourSpecies/errors/firstetraining.stderr - contains errors from first iteration of training AUGUSTUS
braker/yourSpecies/errors/secondetraining.stderr - contains errors from second iteration of training AUGUSTUS
braker/yourSpecies/errors/optimize_augustus.stderr - contains errors optimize_augustus.pl (additional training set for AUGUSTUS)
braker/yourSpecies/errors/augustus*.stderr - contain AUGUSTUS execution errors
braker/yourSpecies/startAlign.stderr - if you provided a protein fasta file, something went wrong during protein alignment
braker/yourSpecies/startAlign.stdout - may give clues on at which point protein alignment went wrong
BRAKER complains that the RNA-Seq file does not correspond to the provided genome file, but I am sure the files correspond to each other!
Please check the headers of the genome FASTA file. If the headers are long and contain whitespaces, some RNA-Seq alignment tools will truncate sequence names in the BAM file. This leads to an error with BRAKER. Solution: shorten/simplify FASTA headers in the genome file before running the RNA-Seq alignment and BRAKER.
GeneMark fails!
(a) GeneMark by default only uses contigs longer than 50k for training. If you have a highly fragmented assembly, this might lead to "no data" for training. You can override the default minimal length by setting the BRAKER argument --min_contig=10000 .
(b) see "[something] failed to execute" below.
[something] failed to execute!
When providing paths to software to BRAKER, please use absolute, non-abbreviated paths. For example, BRAKER might have problems with --SAMTOOLS_PATH=./samtools/ or --SAMTOOLS_PATH=~/samtools/ . Please use SAMTOOLS_PATH=/full/absolute/path/to/samtools/ , instead. This applies to all path specifications as command line options to braker.pl . Relative paths and absolute paths will not pose problems if you export a bash variable, instead, or if you append the location of tools to your $PATH variable.
GeneMark-ETP in BRAKER dies with '/scratch/11232323': No such file or directory.
This appears to be related to sorting large files, and it's a system configuration depending problem. Solve it with export TMPDIR=/tmp/ before calling BRAKER via Singularity.
BRAKER cannot find the Augustus script XYZ...
Update Augustus from github with git clone https://github.com/Gaius-Augustus/Augustus.git . Do not use Augustus from other sources. BRAKER is highly dependent on an up-to-date Augustus. Augustus releases happen rather rarely, updates to the Augustus scripts folder occur rather frequently.
Does BRAKER depend on Python3?
It does. The python scripts employed by BRAKER are not compatible with Python2.
Why does BRAKER predict more genes than I expected?
If transposable elements (or similar) have not been masked appropriately, AUGUSTUS tends to predict those elements as protein coding genes. This can lead to a huge number genes. You can check whether this is the case for your project by BLASTing (or DIAMONDing) the predicted protein sequences against themselves (all vs. all) and counting how many of the proteins have a high number of high quality matches. You can use the output of this analysis to divide your gene set into two groups: the protein coding genes that you want to find and the repetitive elements that were additionally predicted.
I am running BRAKER in Anaconda and something fails...
Update AUGUSTUS and BRAKER from github with git clone https://github.com/Gaius-Augustus/Augustus.git and git clone https://github.com/Gaius-Augustus/BRAKER.git . The Anaconda installation is great, but it relies on releases of AUGUSTUS and BRAKER - which are often lagging behind. Please use the current GitHub code, instead.
Why and where is the GenomeThreader support gone?
BRAKER is a joint project between teams from University of Greifswald and Georgia Tech. While the group of Mark Bordovsky from Georgia Tech contributes GeneMark expertise, the group of Mario Stanke from University of Greifswald contributes AUGUSTUS expertise. Using GenomeThreader to build training genes for AUGUSTUS in BRAKER circumvents execution of GeneMark. Thus, the GenomeThreader mode is strictly speaking not part of the BRAKER project. The previous functionality of BRAKER with GenomeThreader has been moved to GALBA at https://github.com/Gaius-Augustus/GALBA. Note that GALBA has also undergone extension for using Miniprot instead of GenomeThreader.
My BRAKER gene set has too many BUSCO duplicates!
AUGUSTUS within BRAKER can predict alternative splicing isoforms. Also the merge of the AUGUSTUS and GeneMark gene set by TSEBRA within BRAKER may result in additional isoforms for a single gene. The BUSCO duplicates usually come from alternative splicing isoforms, ie they are expected.
Augustus and/or etraining within BRAKER complain that the file aug_cmdln_parameters.json is missing. Even though I am using the latest Singularity container!
BRAKER copies the AUGUSTUS_CONFIG_PATH folder to a writable location. In older versions of Augustus, that file was indeed not existing. If the local writable copy of a folder already exists, BRAKER will not re-copy it. Simply delete the old folder. (It is often ~/.augustus , so you can simply do rm -rf ~/.augustus ; the folder might be residing in $PWD if your home directory was not writable).
I sit behind a firewall, compleasm cannot download the BUSCO files, what can I do? See Issue #785 (comment)
Since BRAKER is a pipeline that calls several Bioinformatics tools, publication of results obtained by BRAKER requires that not only BRAKER is cited, but also the tools that are called by BRAKER. BRAKER will output a file what-to-cite.txt in the BRAKER working directory, informing you about which exact sources apply to your run.
Always cite:
Stanke, M., Diekhans, M., Baertsch, R. and Haussler, D. (2008). Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics, doi: 10.1093/bioinformatics/btn013.
Stanke. M., Schöffmann, O., Morgenstern, B. and Waack, S. (2006). Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics 7, 62.
If you provided any kind of evidence for BRAKER, cite:
If you provided both short read RNA-Seq evidence and a large database of proteins, cite:
Gabriel, L., Bruna, T., Hoff, KJ, Ebel, M., Lomsadze, A., Borodovsky, M., Stanke, M. (2023). BRAKER3: Fully Automated Genome Annotation Using RNA-Seq and Protein Evidence with GeneMark-ETP, AUGUSTUS and TSEBRA. bioRxiV, doi: 10.1101/2023.06.10.54444910.1101/2023.01.01.474747.
Bruna, T., Lomsadze, A., Borodovsky, M. (2023). GeneMark-ETP: Automatic Gene Finding in Eukaryotic Genomes in Consistence with Extrinsic Data. bioRxiv, doi: 10.1101/2023.01.13.524024.
Kovaka, S., Zimin, AV, Pertea, GM, Razaghi, R., Salzberg, SL, & Pertea, M. (2019). Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome biology, 20(1):1-13.
Pertea, G., & Pertea, M. (2020). GFF utilities: GffRead and GffCompare. F1000Research, 9.
Quinlan, AR (2014). BEDTools: the Swiss‐army tool for genome feature analysis. Current protocols in bioinformatics, 47(1):11-12.
If the only source of evidence for BRAKER was a large database of protein sequences, cite:
If the only source of evidence for BRAKER was RNA-Seq data, cite:
Hoff, KJ, Lange, S., Lomsadze, A., Borodovsky, M. and Stanke, M. (2016). BRAKER1: unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics, 32(5):767-769.
Lomsadze, A., Paul DB, and Mark B. (2014) Integration of Mapped Rna-Seq Reads into Automatic Training of Eukaryotic Gene Finding Algorithm. Nucleic Acids Research 42(15): e119--e119
If you called BRAKER3 with an IsoSeq BAM file, or if you envoked the --busco_lineage option, cite:
If you called BRAKER with the --busco_lineage option, in addition, cite:
Simão, FA, Waterhouse, RM, Ioannidis, P., Kriventseva, EV, & Zdobnov, EM (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics, 31(19), 3210-3212.
Li, H. (2023). Protein-to-genome alignment with miniprot. Bioinformatics, 39(1), btad014.
Huang, N., & Li, H. (2023). compleasm: a faster and more accurate reimplementation of BUSCO. Bioinformatics, 39(10), btad595.
If any kind of AUGUSTUS training was performed by BRAKER, check carefully whether you configured BRAKER to use NCBI BLAST or DIAMOND. One of them was used to filter out redundant training gene structures.
If you used NCBI BLAST, please cite:
Altschul, AF, Gish, W., Miller, W., Myers, EW and Lipman, DJ (1990). A basic local alignment search tool. J Mol Biol 215:403--410.
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., and Madden, TL (2009). Blast+: architecture and applications. BMC bioinformatics, 10(1):421.
If you used DIAMOND, please cite:
If BRAKER was executed with a genome file and no extrinsic evidence, cite, then GeneMark-ES was used, cite:
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, YO and Borodovsky, M. (2005). Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Research, 33(20):6494--6506.
Ter-Hovhannisyan, V., Lomsadze, A., Chernoff, YO and Borodovsky, M. (2008). Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome research, pages gr--081612, 2008.
Hoff, KJ, Lomsadze, A., Borodovsky, M. and Stanke, M. (2019). Whole-Genome Annotation with BRAKER. Methods Mol Biol. 1962:65-95, doi: 10.1007/978-1-4939-9173-0_5.
If BRAKER was run with proteins as source of evidence, please cite all tools that are used by the ProtHint pipeline to generate hints:
Bruna, T., Lomsadze, A., & Borodovsky, M. (2020). GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genomics and Bioinformatics, 2(2), lqaa026.
Buchfink, B., Xie, C., Huson, DH (2015). Fast and sensitive protein alignment using DIAMOND. Nature Methods 12:59-60.
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, YO and Borodovsky, M. (2005). Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Research, 33(20):6494--6506.
Iwata, H., and Gotoh, O. (2012). Benchmarking spliced alignment programs including Spaln2, an extended version of Spaln that incorporates additional species-specific features. Nucleic acids research, 40(20), e161-e161.
Gotoh, O., Morita, M., Nelson, DR (2014). Assessment and refinement of eukaryotic gene structure prediction with gene-structure-aware multiple protein sequence alignment. BMC bioinformatics, 15(1), 189.
If BRAKER was executed with RNA-Seq alignments in bam-format, then SAMtools was used, cite:
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R.; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25(16):2078-9.
Barnett, DW, Garrison, EK, Quinlan, AR, Strömberg, MP and Marth GT (2011). BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics, 27(12):1691-2
If BRAKER downloaded RNA-Seq libraries from SRA using their IDs, cite SRA, SRA toolkit, and HISAT2:
Leinonen, R., Sugawara, H., Shumway, M., & International Nucleotide Sequence Database Collaboration. (2010). The sequence read archive. Nucleic acids research, 39(suppl_1), D19-D21.
SRA Toolkit Development Team (2020). SRA Toolkit. https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software.
Kim, D., Paggi, JM, Park, C., Bennett, C., & Salzberg, SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature biotechnology, 37(8):907-915.
If BRAKER was executed using RNA-Seq data in FASTQ format, cite HISAT2:
If BRAKER called MakeHub for creating a track data hub for visualization of BRAKER results with the UCSC Genome Browser, cite:
If BRAKER called GUSHR for generating UTRs, cite:
Keilwagen, J., Hartung, F., Grau, J. (2019) GeMoMa: Homology-based gene prediction utilizing intron position conservation and RNA-seq data. Methods Mol Biol. 1962:161-177, doi: 10.1007/978-1-4939-9173-0_9.
Keilwagen, J., Wenk, M., Erickson, JL, Schattat, MH, Grau, J., Hartung F. (2016) Using intron position conservation for homology-based gene prediction. Nucleic Acids Research, 44(9):e89.
Keilwagen, J., Hartung, F., Paulini, M., Twardziok, SO, Grau, J. (2018) Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinformatics, 19(1):189.
All source code, ie scripts/*.pl or scripts/*.py are under the Artistic License (see http://www.opensource.org/licenses/artistic-license.php).
[F1] EX = ES/ET/EP/ETP, all available for download under the name GeneMark-ES/ET/EP ↩
[F2] Please use the latest version from the master branch of AUGUSTUS distributed by the original developers, it is available from github at https://github.com/Gaius-Augustus/Augustus. Problems have been reported from users that tried to run BRAKER with AUGUSTUS releases maintained by third parties, ie Bioconda. ↩
[F4] install with sudo apt-get install cpanminus ↩
[F6] The binary may eg reside in bamtools/build/src/toolkit ↩
[R0] Bruna, Tomas, Hoff, Katharina J., Lomsadze, Alexandre, Stanke, Mario, and Borodovsky, Mark. 2021. “BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database." NAR Genomics and Bioinformatics 3(1):lqaa108.↩
[R1] Hoff, Katharina J, Simone Lange, Alexandre Lomsadze, Mark Borodovsky, and Mario Stanke. 2015. “BRAKER1: Unsupervised Rna-Seq-Based Genome Annotation with Genemark-et and Augustus.” Bioinformatics 32 (5). Oxford University Press: 767--69.↩
[R2] Lomsadze, Alexandre, Paul D Burns, and Mark Borodovsky. 2014. “Integration of Mapped Rna-Seq Reads into Automatic Training of Eukaryotic Gene Finding Algorithm.” Nucleic Acids Research 42 (15). Oxford University Press: e119--e119.↩
[R3] Stanke, Mario, Mark Diekhans, Robert Baertsch, and David Haussler. 2008. “Using Native and Syntenically Mapped cDNA Alignments to Improve de Novo Gene Finding.” Bioinformatics 24 (5). Oxford University Press: 637--44.↩
[R4] Stanke, Mario, Oliver Schöffmann, Burkhard Morgenstern, and Stephan Waack. 2006. “Gene Prediction in Eukaryotes with a Generalized Hidden Markov Model That Uses Hints from External Sources.” BMC Bioinformatics 7 (1). BioMed Central: 62.↩
[R5] Barnett, Derek W, Erik K Garrison, Aaron R Quinlan, Michael P Strömberg, and Gabor T Marth. 2011. “BamTools: A C++ Api and Toolkit for Analyzing and Managing Bam Files.” Bioinformatics 27 (12). Oxford University Press: 1691--2.↩
[R6] Li, Heng, Handsaker, Bob, Alec Wysoker, Tim Fennell, Jue Ruan, Nils Homer, Gabor Marth, Goncalo Abecasis, and Richard Durbin. 2009. “The Sequence Alignment/Map Format and Samtools.” Bioinformatics 25 (16). Oxford University Press: 2078--9.↩
[R7] Gremme, G. 2013. “Computational Gene Structure Prediction.” PhD thesis, Universität Hamburg.↩
[R8] Gotoh, Osamu. 2008a. “A Space-Efficient and Accurate Method for Mapping and Aligning cDNA Sequences onto Genomic Sequence.” Nucleic Acids Research 36 (8). Oxford University Press: 2630--8.↩
[R9] Iwata, Hiroaki, and Osamu Gotoh. 2012. “Benchmarking Spliced Alignment Programs Including Spaln2, an Extended Version of Spaln That Incorporates Additional Species-Specific Features.” Nucleic Acids Research 40 (20). Oxford University Press: e161--e161.↩
[R10] Osamu Gotoh. 2008b. “Direct Mapping and Alignment of Protein Sequences onto Genomic Sequence.” Bioinformatics 24 (21). Oxford University Press: 2438--44.↩
[R11] Slater, Guy St C, and Ewan Birney. 2005. “Automated Generation of Heuristics for Biological Sequence Comparison.” BMC Bioinformatics 6(1). BioMed Central: 31.↩
[R12] Altschul, SF, W. Gish, W. Miller, EW Myers, and DJ Lipman. 1990. “Basic Local Alignment Search Tool.” Journal of Molecular Biology 215:403--10.↩
[R13] Camacho, Christiam, et al. 2009. “BLAST+: architecture and applications.“ BMC Bioinformatics 1(1): 421.↩
[R14] Lomsadze, A., V. Ter-Hovhannisyan, YO Chernoff, and M. Borodovsky. 2005. “Gene identification in novel eukaryotic genomes by self-training algorithm.” Nucleic Acids Research 33 (20): 6494--6506. doi:10.1093/nar/gki937.↩
[R15] Ter-Hovhannisyan, Vardges, Alexandre Lomsadze, Yury O Chernoff, and Mark Borodovsky. 2008. “Gene Prediction in Novel Fungal Genomes Using an Ab Initio Algorithm with Unsupervised Training.” Genome Research . Cold Spring Harbor Lab, gr--081612.↩
[R16] Hoff, KJ 2019. MakeHub: Fully automated generation of UCSC Genome Browser Assembly Hubs. Genomics, Proteomics and Bioinformatics , in press, preprint on bioarXive, doi: https://doi.org/10.1101/550145.↩
[R17] Bruna, T., Lomsadze, A., & Borodovsky, M. 2020. GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genomics and Bioinformatics, 2(2), lqaa026. doi: https://doi.org/10.1093/nargab/lqaa026.↩
[R18] Kriventseva, EV, Kuznetsov, D., Tegenfeldt, F., Manni, M., Dias, R., Simão, FA, and Zdobnov, EM 2019. OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Research, 47(D1), D807-D811.↩
[R19] Keilwagen, J., Hartung, F., Grau, J. (2019) GeMoMa: Homology-based gene prediction utilizing intron position conservation and RNA-seq data. Methods Mol Biol. 1962:161-177, doi: 10.1007/978-1-4939-9173-0_9.↩
[R20] Keilwagen, J., Wenk, M., Erickson, JL, Schattat, MH, Grau, J., Hartung F. (2016) Using intron position conservation for homology-based gene prediction. Nucleic Acids Research, 44(9):e89.↩
[R21] Keilwagen, J., Hartung, F., Paulini, M., Twardziok, SO, Grau, J. (2018) Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinformatics, 19(1):189.↩
[R22] SRA Toolkit Development Team (2020). SRA Toolkit. https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software.[↩](#a22)
[R23] Kim, D., Paggi, JM, Park, C., Bennett, C., & Salzberg, SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature biotechnology, 37(8):907-915.↩
[R24] Quinlan, AR (2014). BEDTools: the Swiss‐army tool for genome feature analysis. Current protocols in bioinformatics, 47(1):11-12.↩
[R25] Kovaka, S., Zimin, AV, Pertea, GM, Razaghi, R., Salzberg, SL, & Pertea, M. (2019). Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome biology, 20(1):1-13.↩
[R26] Pertea, G., & Pertea, M. (2020). GFF utilities: GffRead and GffCompare. F1000Research, 9.↩
[R27] Huang, N., & Li, H. (2023). compleasm: a faster and more accurate reimplementation of BUSCO. Bioinformatics, 39(10), btad595.↩
[R28] Bruna, T., Gabriel, L. & Hoff, KJ (2024). Navigating Eukaryotic Genome Annotation Pipelines: A Route Map to BRAKER, Galba, and TSEBRA. arXiv, https://doi.org/10.48550/arXiv.2403.19416 .↩


