This repo contains:
sepal requires python3, preferably a version later than or equal to 3.5. To
download and install, open the terminal and change to a directory where you want
sepal to be downloaded to and do:
git clone https://github.com/almaan/sepal.git
cd sepal
chmod +x setup.py
./setup.py install
Depending on your user privileges, you may have to add --user as an argument to setup.py.
Running the setup will give you the minimal required install to compute the diffusion times. However,
if you want to be able to use the analysis modules, you also need to install the recommended packages.
To do this, simply (in the same directory) run:
pip install -e ".[full]"again, the --user could be necessary to include. Also, you may have to use pip3 if this is the way you've set up your python-pip interface. If your using conda or virtual environments, follow their recommendations for installation of packages.
This should install both a command line interface (CLI) and a standard package. To test and see whether the installation was successful you could try executing the command:
sepal -h
Which should print the help message associated with sepal. If everything worked out for you so far,
you may proceed to the example section to see sepal in action!
The recommended usage of sepal is by
the command line interface. Both the simulations
in order to compute the diffusion times as well
as subsequent analysis or inspection of the results
can easily be performed by typing sepal followed by
either run or analyze. The analyze module has different
options, to visualize the results (inspect),
sort the profiles into pattern families (family) or subject
the identified families to functional enrichment analysis (fea). For a
complete list of commands available, do sepal module -h, where module
is one of run and analyze. Below, we illustrate
how sepal may be used to find transcription profiles with spatial patterns.
We will create a folder to hold our results, which will also figure as our working directory. From the main directory of the repo, do:
cd res
mkdir example
cd exampleThe MOB sample will be used to exemplify our analysis. We begin with calculating diffusion times for each transcription profile:
sepal run -c ../../data/real/mob.tsv.gz -mo 10 -mc 5 -o . -ar 1Below is an example (with an additional display of the help command) of how this might look

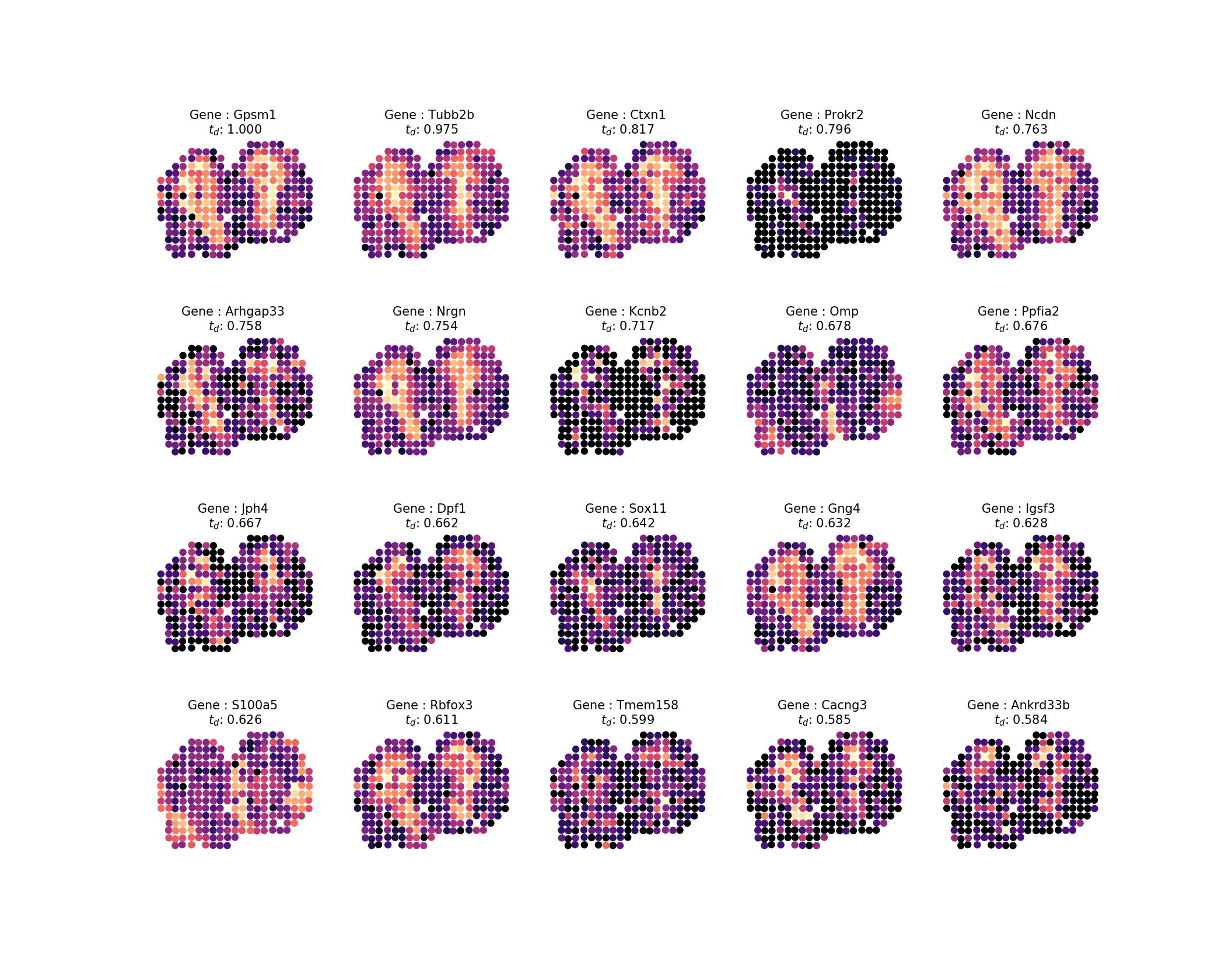
Having computed the diffusion times, we want to inspect the result, like in the study, we will look at the top 20 profiles. We can easily generate images from our result by running the command:
sepal analyze -c ../../data/real/mob.tsv.gz
-r 20200409173043610345-top-diffusion-times.tsv
-ar 1k -o . inspect -ng 20 -nc 5Which would look something in the line of this:

The output wil be the following image:
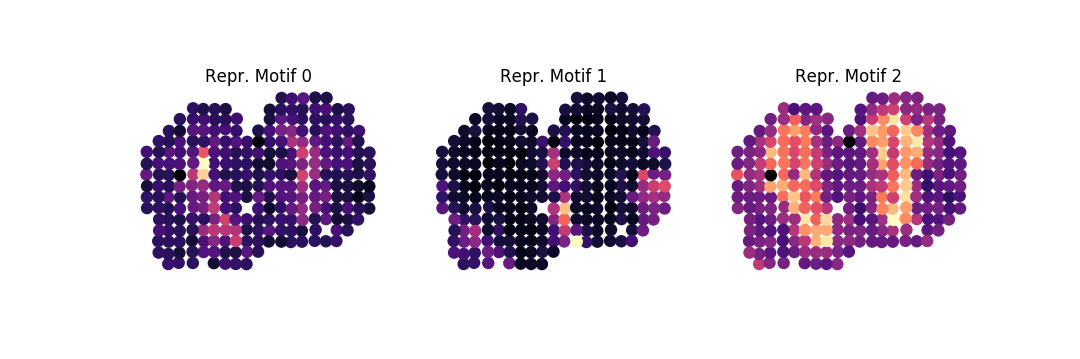
Then, to sort the 100 top ranked genes into a set of pattern families, where 85% of the variance in our patterns should be explained by the eigenpatterns, do:
sepal analyze -c ../../data/real/mob.tsv.gz
-r 20200409173043610345-top-diffusion-times.tsv
-ar 1k -o . family -ng 100 -nbg 100 -eps 0.85 --plot -nc 3From this, we obtain the following three representative motifs for each family:
We may subject our families to enrichment analysis, by running:
sepal analyze -c ../../data/real/mob.tsv.gz
-r 20200409173043610345-top-diffusion-times.tsv
-ar 1k -o . fea -fl mob.tsv-family-index.tsv -or "mmusculus"where we for example see that Family 2 is enriched for several processes related to neuronal function, generation and regulation:
| family | native | name | p_value | source | intersection_size | |
|---|---|---|---|---|---|---|
| 2 | 2 | GO:0007399 | nervous system development | 0.00035977 | GO:BP | 26 |
| 3 | 2 | GO:0050773 | regulation of dendrite development | 0.000835883 | GO:BP | 8 |
| 4 | 2 | GO:0048167 | regulation of synaptic plasticity | 0.00196494 | GO:BP | 8 |
| 5 | 2 | GO:0016358 | dendrite development | 0.00217167 | GO:BP | 9 |
| 6 | 2 | GO:0048813 | dendrite morphogenesis | 0.00741589 | GO:BP | 7 |
| 7 | 2 | GO:0048814 | regulation of dendrite morphogenesis | 0.00800399 | GO:BP | 6 |
| 8 | 2 | GO:0048666 | neuron development | 0.0114088 | GO:BP | 16 |
| 9 | 2 | GO:0099004 | calmodulin dependent kinase signaling pathway | 0.0159572 | GO:BP | 3 |
| 10 | 2 | GO:0050804 | modulation of chemical synaptic transmission | 0.0341913 | GO:BP | 10 |
| 11 | 2 | GO:0099177 | regulation of trans-synaptic signaling | 0.0347783 | GO:BP | 10 |
Of course, this analysis is by no means exhaustive. But rather an quick example to show how one operates the CLI for sepal .
While sepal has been designed as a standalone tool, we've also constructed it
to be functional as a standard python package from which functions may be
imported and used in an integrated workflow. To show how this may be done, we
provide an example, reproducing the melanoma analysis. More examples may be added later on.
The input to sepal is required to be in the format n_locations x n_genes,
however if your data is structured in the opposite way (n_genes x n_locations)
simply provide the --transpose flag when running either the simulation or
analysis and this will be taken care of.
We currently support .csv,.tsv and .h5ad formats. For the latter, your file should be structured
according to THIS format. We expect that there will be a release from the
scanpy team in the near future, where a standardized format for spatial data is presented, but until then we will be
using the aforementioned standard.
All real data we used is public, and can be found accessed at the following links:
The synthetic data was generated by:
synthetic/img2cnt.py
synthetic/turing.py
synthetic/ablation.py
All the results presented in the study can be found in the res folder, both for
the real and synthetic data. For each sample we have structured the results accordingly:
res/
sample-name/
X-diffusion-times.tsv : diffusion times for all ranked genesanalysis/ : contains output of secondary analysis

