Este repositorio contiene:
sepal requiere python3 , preferiblemente una versión posterior o igual a 3.5. Para descargar e instalar, abra la terminal y cambie a un directorio donde desea descargar sepal y haga:
git clone https://github.com/almaan/sepal.git
cd sepal
chmod +x setup.py
./setup.py install
Dependiendo de sus privilegios de usuario, es posible que deba agregar --user como argumento a setup.py . Ejecutar la configuración le brindará la instalación mínima requerida para calcular los tiempos de difusión. Sin embargo, si desea poder utilizar los módulos de análisis, también deberá instalar los paquetes recomendados. Para hacer esto, simplemente (en el mismo directorio) ejecute:
pip install -e " .[full] " Nuevamente, podría ser necesario incluir --user . Además, es posible que tengas que usar pip3 si esta es la forma en que configuraste tu interfaz python-pip . Si utiliza conda o entornos virtuales, siga sus recomendaciones para la instalación de paquetes.
Esto debería instalar tanto una interfaz de línea de comandos (CLI) como un paquete estándar. Para probar y ver si la instalación fue exitosa, puede intentar ejecutar el comando:
sepal -h
Que debería imprimir el mensaje de ayuda asociado con el sépalo. Si todo funcionó para usted hasta ahora, puede continuar con la sección de ejemplos para ver sepal en acción.
El uso recomendado de sépal es mediante la interfaz de línea de comandos. Tanto las simulaciones para calcular los tiempos de difusión como el análisis o inspección posterior de los resultados se pueden realizar fácilmente escribiendo sepal seguido de run o analyze . El módulo analyze tiene diferentes opciones, para visualizar los resultados ( inspect ), ordenar los perfiles en familias de patrones ( family ) o someter las familias identificadas a un análisis de enriquecimiento funcional ( fea ). Para obtener una lista completa de los comandos disponibles, ejecute sepal module -h , donde módulo es uno de run y analyze . A continuación, ilustramos cómo se puede utilizar el sépalo para encontrar perfiles de transcripción con patrones espaciales.
Crearemos una carpeta para guardar nuestros resultados, que también figurará como nuestro directorio de trabajo. Desde el directorio principal del repositorio, haga:
cd res
mkdir example
cd exampleLa muestra MOB se utilizará para ejemplificar nuestro análisis. Comenzamos calculando los tiempos de difusión para cada perfil de transcripción:
sepal run -c ../../data/real/mob.tsv.gz -mo 10 -mc 5 -o . -ar 1A continuación se muestra un ejemplo (con una visualización adicional del comando de ayuda) de cómo podría verse esto.

Habiendo calculado los tiempos de difusión, queremos inspeccionar el resultado, como en el estudio, veremos los 20 perfiles principales. Podemos generar fácilmente imágenes a partir de nuestro resultado ejecutando el comando:
sepal analyze -c ../../data/real/mob.tsv.gz
-r 20200409173043610345-top-diffusion-times.tsv
-ar 1k -o . inspect -ng 20 -nc 5Lo cual se vería algo en la línea de esto:

El resultado será la siguiente imagen:
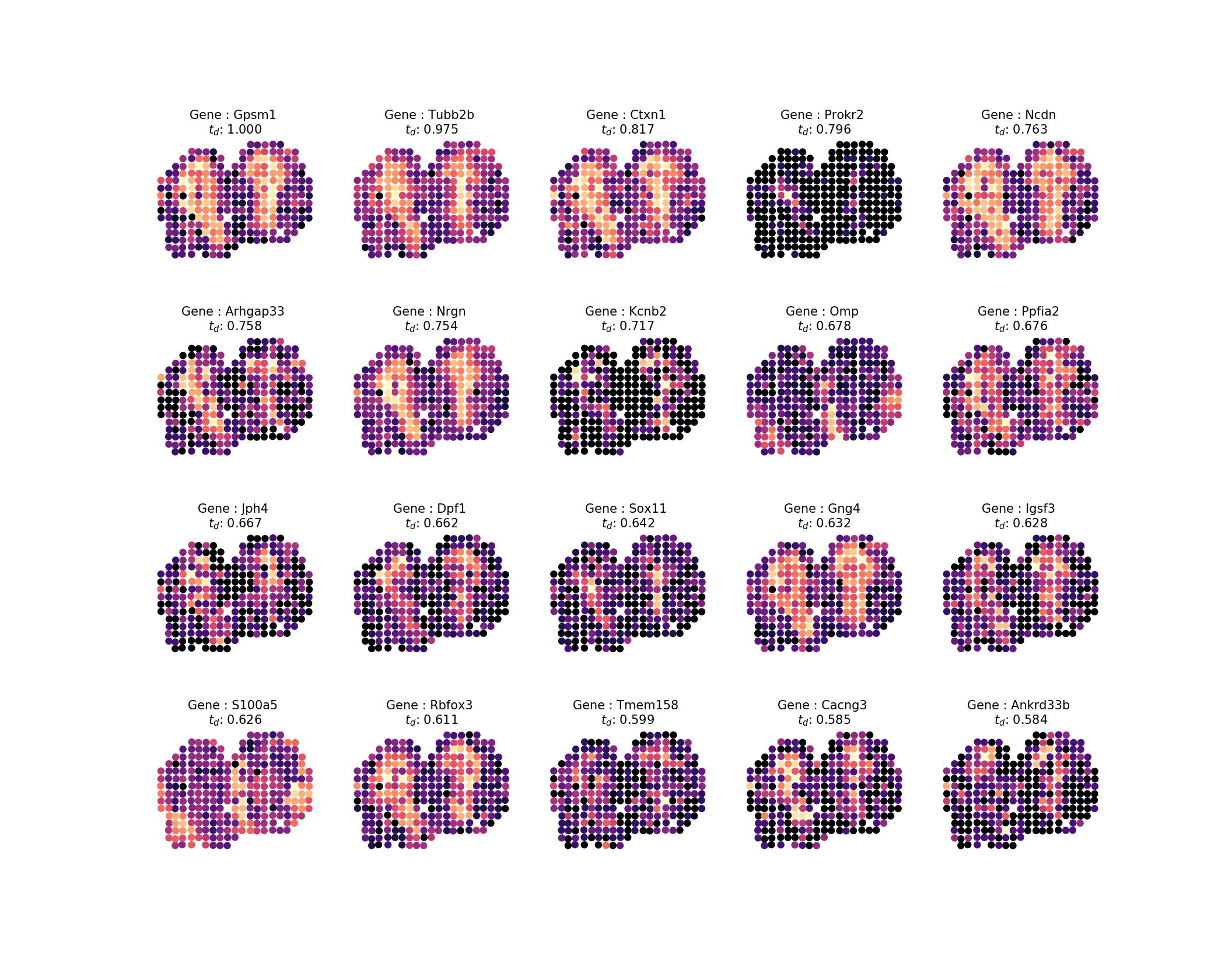
Luego, para clasificar los 100 genes mejor clasificados en un conjunto de familias de patrones, donde el 85% de la varianza en nuestros patrones debería explicarse por los patrones propios, haga lo siguiente:
sepal analyze -c ../../data/real/mob.tsv.gz
-r 20200409173043610345-top-diffusion-times.tsv
-ar 1k -o . family -ng 100 -nbg 100 -eps 0.85 --plot -nc 3De aquí obtenemos los siguientes tres motivos representativos de cada familia:
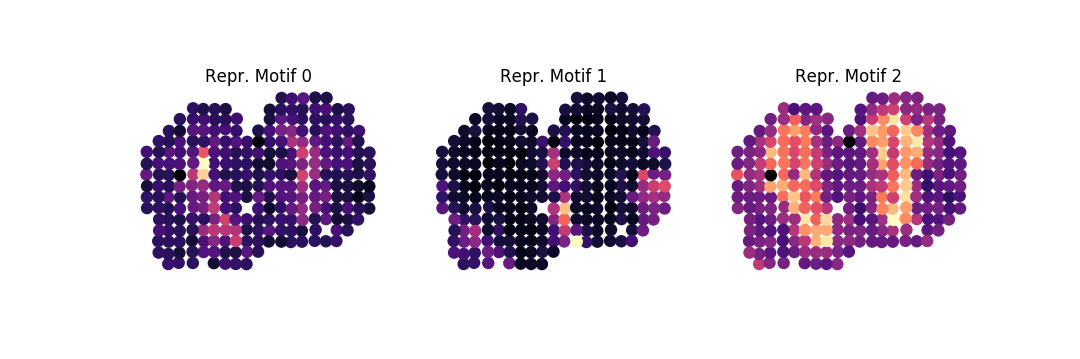
Podemos someter a nuestras familias a un análisis de enriquecimiento, ejecutando:
sepal analyze -c ../../data/real/mob.tsv.gz
-r 20200409173043610345-top-diffusion-times.tsv
-ar 1k -o . fea -fl mob.tsv-family-index.tsv -or " mmusculus "donde vemos por ejemplo que la Familia 2 está enriquecida para varios procesos relacionados con la función, generación y regulación neuronal:
| familia | nativo | nombre | valor_p | fuente | tamaño_intersección | |
|---|---|---|---|---|---|---|
| 2 | 2 | IR: 0007399 | desarrollo del sistema nervioso | 0.00035977 | IR: BP | 26 |
| 3 | 2 | IR: 0050773 | regulación del desarrollo de las dendritas | 0.000835883 | IR: BP | 8 |
| 4 | 2 | IR: 0048167 | regulación de la plasticidad sináptica | 0.00196494 | IR: BP | 8 |
| 5 | 2 | IR: 0016358 | desarrollo dendrítico | 0.00217167 | IR: BP | 9 |
| 6 | 2 | IR: 0048813 | morfogénesis dendrítica | 0.00741589 | IR: BP | 7 |
| 7 | 2 | IR: 0048814 | regulación de la morfogénesis de las dendritas | 0.00800399 | IR: BP | 6 |
| 8 | 2 | IR: 0048666 | desarrollo neuronal | 0.0114088 | IR: BP | 16 |
| 9 | 2 | IR:0099004 | vía de señalización de la quinasa dependiente de calmodulina | 0.0159572 | IR: BP | 3 |
| 10 | 2 | IR: 0050804 | Modulación de la transmisión sináptica química. | 0.0341913 | IR: BP | 10 |
| 11 | 2 | IR: 0099177 | regulación de la señalización transináptica | 0.0347783 | IR: BP | 10 |
Por supuesto, este análisis no es en modo alguno exhaustivo. Pero más bien un ejemplo rápido para mostrar cómo se opera la CLI para sepal .
Si bien sepal ha sido diseñado como una herramienta independiente, también lo hemos construido para que funcione como un paquete estándar de Python desde el cual se pueden importar funciones y utilizarlas en un flujo de trabajo integrado. Para mostrar cómo se puede hacer esto, proporcionamos un ejemplo que reproduce el análisis del melanoma. Es posible que se agreguen más ejemplos más adelante.
Se requiere que la entrada a sepal esté en el formato n_locations x n_genes ; sin embargo, si sus datos están estructurados de manera opuesta ( n_genes x n_locations ), simplemente proporcione el indicador --transpose cuando ejecute la simulación o el análisis y esto se solucionará. de.
Actualmente admitimos los formatos .csv , .tsv y .h5ad . Para este último, su archivo debe estructurarse según ESTE formato. Esperamos que en un futuro cercano haya un lanzamiento del equipo scanpy , donde se presente un formato estandarizado para datos espaciales, pero hasta entonces usaremos el estándar antes mencionado.
Todos los datos reales que utilizamos son públicos y se pueden consultar en los siguientes enlaces:
Los datos sintéticos fueron generados por:
synthetic/img2cnt.pysynthetic/turing.pysynthetic/ablation.py Todos los resultados presentados en el estudio se pueden encontrar en la carpeta res , tanto para los datos reales como para los sintéticos. Para cada muestra hemos estructurado los resultados en consecuencia:
res/sample-name/X-diffusion-times.tsv : tiempos de difusión para todos los genes clasificadosanalysis/ : contiene resultados del análisis secundario

