reference_database_creator
bug fix --in-silico-pcr --untrimmed
CANGREJOS ( do comer R bases de datos de referencia para A mplicón- B asado S equencing) es un programa de software versátil que genera bases de datos de referencia seleccionadas para análisis metagenómicos. El flujo de trabajo de CRABS consta de siete módulos: (i) descargar datos de repositorios en línea; (ii) importar datos descargados al formato CRABS; (iii) extraer regiones de amplicones mediante análisis de PCR in silico ; (iv) recuperar amplicones sin regiones de unión a cebadores mediante alineamientos con códigos de barras extraídos in silico ; (v) seleccionar y crear subconjuntos de la base de datos local mediante múltiples parámetros de filtrado; (vi) exportar la base de datos local en varios formatos de acuerdo con los requisitos del clasificador taxonómico; y (vi) funciones de posprocesamiento, es decir, visualizaciones, para explorar y proporcionar una descripción resumida de la base de datos de referencia local. Estos siete módulos se dividen en dieciocho funciones y se describen a continuación. Además, se proporciona código de ejemplo para cada una de las dieciocho funciones. Finalmente, al final de este documento README se proporciona un tutorial para crear una base de datos de referencia de tiburones local para el conjunto de cebadores MiFish-E para proporcionar un script de ejemplo como referencia.
Nos complace anunciar que CRABS ha experimentado una importante actualización y un rediseño del código basado en los comentarios de los usuarios, que esperamos mejore la experiencia del usuario al crear su propia base de datos de referencia local.
A continuación encontrará una lista de características y mejoras agregadas a CRABS v 1.0.0 :
CRABS v 1.0.0 ahora se puede descargar manualmente clonando este repositorio de GitHub (consulte 4.1 Instalación manual para obtener información detallada). Actualizaremos el contenedor Docker y el paquete conda lo antes posible para facilitar la instalación de la versión más reciente.
Cuando utilice CRABS en sus proyectos de investigación, cite el siguiente artículo:
[Jeunen, G.-J., Dowle, E., Edgecombe, J., von Ammon, U., Gemmell, N. J., & Cross, H. (2022). crabs—A software program to generate curated reference databases for metabarcoding sequencing data. Molecular Ecology Resources, 00, 1– 14.](https://doi.org/10.1111/1755-0998.13741)
CRABS es un conjunto de herramientas de línea de comandos que se ejecuta en entornos típicos de Unix/Linux y está escrito exclusivamente en python3. Sin embargo, CRABS utiliza el módulo de subproceso en Python para ejecutar varios comandos en sintaxis bash para evitar las idiosincrasias específicas de Python y aumentar la velocidad de ejecución. Ofrecemos tres formas de instalar CRABS. Para la versión más actualizada de CRABS, recomendamos la instalación manual clonando este repositorio de GitHub e instalando 10 dependencias por separado (las instrucciones de instalación para todas las dependencias se proporcionan en 4.1 Instalación manual). CRABS también se puede instalar mediante Docker y conda. Ambos métodos permiten una fácil instalación al coinstalar automáticamente todas las dependencias. Nuestro objetivo es mantener actualizados el contenedor Docker y el paquete conda, aunque puede producirse un cierto retraso en la actualización a la versión más reciente, especialmente para el paquete conda. A continuación se detallan los tres enfoques.
Para la instalación manual, primero clone el repositorio CRABS. Este paso requiere que GitHub esté disponible en la línea de comandos (instrucciones de instalación para GitHub).
git clone https://github.com/gjeunen/reference_database_creator.git
Dependiendo de su configuración, es posible que CRABS deba hacerse ejecutable en su sistema. Esto se puede lograr usando el siguiente código.
chmod +x reference_database_creator/crabs
Una vez instalado CRABS, debemos asegurarnos de que todas las dependencias estén instaladas y sean accesibles globalmente. La última versión de CRABS (versión v 1.0.0 ) se ejecuta en Python 3.11.7 (o cualquier versión compatible con 3.11.7) y se basa en cinco módulos de Python que pueden no venir de serie con Python, así como en cinco programas de software externos. Todas las dependencias se enumeran a continuación, junto con un enlace a las instrucciones de instalación. Los números de versión proporcionados para cada módulo y programa de software son aquellos en los que se desarrolló CRABS. Aunque también se podrían utilizar versiones compatibles de cada uno.
Módulos de Python:
Programas de software externos:
Una vez instalados CRABS y todas las dependencias, se puede acceder a CRABS en todo el sistema operativo utilizando el siguiente código.
export PATH="/path/to/crabs/folder:$PATH"
Sustituya /ruta/a/crabs/carpeta con la ruta real a la carpeta del repositorio de GitHub en el sistema operativo, es decir, la carpeta creada durante el comando git clone anterior. Agregar el código export al archivo .bash_profile o .bashrc hará que CRABS sea accesible globalmente en cualquier momento.
Docker es un proyecto de código abierto que permite la implementación de aplicaciones de software dentro de "contenedores" que están aislados de su computadora y se ejecutan a través de un sistema operativo de host virtual llamado Docker Engine. La principal ventaja de ejecutar Docker en máquinas virtuales es que utilizan muchos menos recursos. Este aislamiento significa que puede ejecutar un contenedor Docker en la mayoría de los sistemas operativos, incluidos Mac, Windows y Linux. Es posible que necesites configurar una cuenta gratuita para utilizar Docker Desktop. Este enlace tiene una buena introducción a los conceptos básicos del uso de Docker. Aquí hay un enlace para comenzar y orientarse en el multiverso Docker.
Sólo hay dos pasos para que Crabs se ejecute en su computadora. Primero, instale Docker Desktop en su computadora, que es gratuito para la mayoría de los usuarios. Aquí están las instrucciones para Mac ; Aquí están las instrucciones para computadoras con Windows y aquí están las instrucciones para Linux (la mayoría de las principales plataformas Linux son compatibles). Una vez que tenga Docker Desktop instalado y ejecutándose (la aplicación de escritorio debe estar ejecutándose para que pueda usar cualquier comando de Docker en la línea de comando), solo tiene que "extraer" nuestra imagen de Crabs y estará listo para comenzar:
docker pull quay.io/swordfish/crabs:0.1.7
Si bien la instalación de una aplicación acoplable es fácil, usar esas aplicaciones puede resultar un poco complicado al principio. Para ayudarlo a comenzar, le proporcionamos algunos comandos de ejemplo utilizando la versión acoplable de cangrejos. Estos ejemplos se pueden encontrar en la carpeta docker_intro de este repositorio . A partir de estos ejemplos, debería poder ejecutar la configuración de una base de datos de referencia completa y estar listo para comenzar. Continuaremos ampliando estos ejemplos y probándolos en muchas situaciones diferentes. Haga preguntas y proporcione comentarios en la pestaña Problemas.
Para instalar el paquete conda, primero debe instalar conda. Vea este enlace para más detalles. Si conda ya está instalado, es una buena práctica actualizar la herramienta conda con conda update conda antes de instalar CRABS.
Una vez instalado conda, siga los pasos a continuación para instalar CRABS y todas las dependencias. Asegúrese de ingresar los comandos en el orden en que aparecen a continuación.
conda create -n CRABS
conda activate CRABS
conda config --add channels bioconda
conda config --add channels conda-forge
conda install -c bioconda crabs
Una vez que haya ingresado el comando de instalación, conda procesará la solicitud (esto puede demorar aproximadamente un minuto) y luego mostrará todos los paquetes y programas que se instalarán y solicitará confirmación. Escriba y para iniciar la instalación. Una vez que esto termine, CANGREJOS deberían estar listos para funcionar.
Hemos probado esta instalación en sistemas Mac y Linux. Aún no lo hemos probado en el subsistema de Windows para Linux (WSL).
Utilice el siguiente código para comprobar si CRABS se instaló correctamente y obtenga la información de ayuda.
crabs -hLa información de ayuda divide las dieciocho funciones en diferentes grupos, y cada grupo enumera la función en la parte superior y los parámetros requeridos y opcionales debajo.

CRABS contiene siete módulos, que incorporan dieciocho funciones:
Módulo 1: descargar datos de repositorios en línea
--download-taxonomy : descarga información de taxonomía NCBI;--download-bold : descarga datos de secuencia de la base de datos Barcode of Life (BOLD);--download-embl : descarga datos de secuencia del Archivo Europeo de Nucleótidos (ENA; EMBL);--download-mitofish : descarga datos de secuencia de la base de datos MitoFish;--download-ncbi : descarga datos de secuencia del Centro Nacional de Información Biotecnológica (NCBI).Módulo 2: importar datos descargados al formato CRABS
--import : importa secuencias descargadas o códigos de barras personalizados al formato CRABS;--merge : fusiona diferentes archivos con formato CRABS en un solo archivo.Módulo 3: extraer regiones de amplicones mediante análisis de PCR in silico
--in-silico-pcr : extrae amplicones de los datos descargados localizando y eliminando regiones de unión de cebadores.Módulo 4: recuperar amplicones sin regiones de unión a cebadores
--pairwise-global-alignment : recupera amplicones sin regiones de unión a cebadores alineando secuencias descargadas con códigos de barras extraídos in silico .Módulo 5: seleccionar y crear subconjuntos de la base de datos local mediante múltiples parámetros de filtrado
--dereplicate : descarta secuencias duplicadas;--filter : descarta secuencias mediante múltiples parámetros de filtrado;--subset : subconjunto de la base de datos local para retener o excluir grupos taxonómicos específicos.Módulo 6: exportar la base de datos local
--export : exporta la base de datos con formato CRABS a varios formatos según los requisitos del clasificador taxonómico a utilizar.Módulo 7: funciones de posprocesamiento para explorar y proporcionar una descripción general resumida de la base de datos de referencia local
--diversity-figure : crea un gráfico de barras horizontales que muestra el número de especies y grupos de secuencias por nivel específico incluidos en la base de datos de referencia;--amplicon-length-figure : crea un gráfico de líneas que representa las distribuciones de longitud de amplicones separadas por grupo taxonómico;--phylogenetic-tree : crea un árbol filogenético con códigos de barras de la base de datos de referencia para una lista de especies objetivo;--amplification-efficiency-figure : crea un gráfico de barras que muestra discrepancias en las regiones de unión del cebador;--completeness-table : crea una hoja de cálculo que contiene la disponibilidad de códigos de barras para grupos taxonómicos.CRABS puede descargar los datos de secuenciación iniciales desde cuatro repositorios en línea, incluidos (i) BOLD, (ii) EMBL, (iii) MitoFish y NCBI. A partir de la versión v 1.0.0 , la descarga de datos de cada repositorio se divide en su propia función. Además, CRABS no formatea automáticamente los datos después de la descarga para aumentar la flexibilidad y permitir la depuración cuando falla la descarga de datos.
Además de descargar datos de secuencia, CRABS también es capaz de descargar la información de taxonomía NCBI, que CRABS utiliza para crear el linaje taxonómico para cada secuencia.
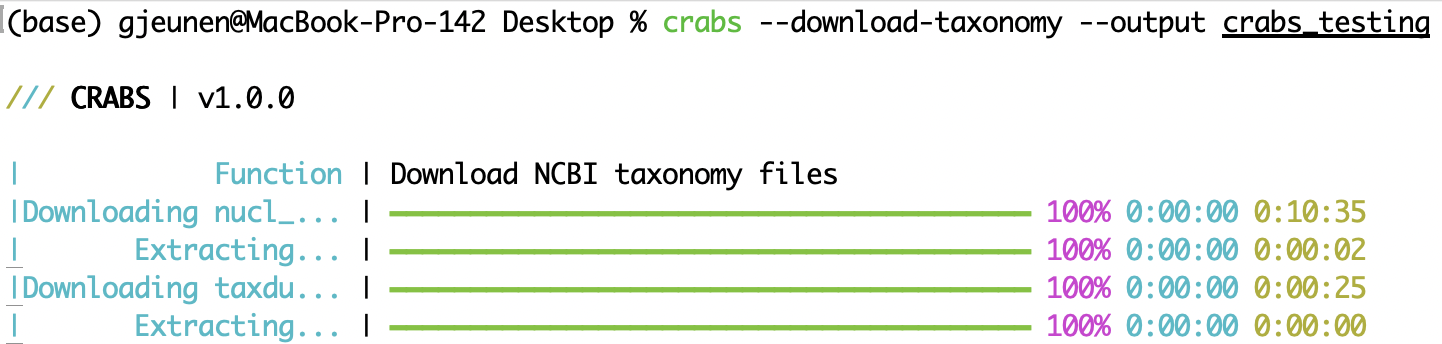
--download-taxonomy Para asignar un linaje taxonómico a cada secuencia descargada en la base de datos de referencia (ver 5.2 Módulo 2), es necesario descargar la información taxonómica. CRABS utiliza la taxonomía del NCBI y descarga tres archivos específicos a su computadora: (i) un archivo que vincula los números de acceso con las identificaciones taxonómicas ( nucl_gb.accession2taxid ), (ii) un archivo que contiene información sobre el nombre filogenético asociado con cada identificación taxonómica ( names.dmp ), y (iii) un archivo que contiene información sobre cómo se vinculan las ID taxonómicas ( nodes.dmp ). El directorio de salida para los archivos descargados se puede especificar utilizando el parámetro --output . Para excluir el archivo nucl_gb.accession2taxid o los archivos nombres.dmp y nodes.dmp , se puede proporcionar el parámetro --exclude acc2tax o --exclude taxdump , respectivamente. El primer código a continuación no descarga ningún archivo, ya que se proporcionan tanto acc2tax como taxdump para el parámetro --exclude . La segunda línea de código descarga los tres archivos al subdirectorio --output crabs_testing . La captura de pantalla siguiente muestra lo que se imprime en la consola al ejecutar esta línea de código.
crabs --download-taxonomy --exclude 'acc2taxid,taxdump'
crabs --download-taxonomy --output crabs_testing
--download-bold Las secuencias BOLD se descargan a través del sitio web BOLD. El archivo de salida, que está estructurado como un documento fasta de dos líneas, se puede especificar utilizando el parámetro --output . Los usuarios pueden especificar qué grupo taxonómico descargar utilizando el parámetro --taxon . Recomendamos escribir un bucle for simple (el ejemplo se proporciona a continuación) cuando los usuarios quieran descargar varios grupos taxonómicos, limitando así la cantidad de datos que se descargarán de BOLD por instancia. Sin embargo, si sólo interesa un número limitado de grupos taxonómicos, los nombres de los grupos taxonómicos también pueden separarse por | (ejemplo proporcionado a continuación). También recomendamos a los usuarios que verifiquen si el nombre del grupo taxonómico que se descargará aparece en el archivo BOLD o si es necesario utilizar nombres alternativos. Por ejemplo, al especificar --taxon Chondrichthyes no se descargarán todas las secuencias de peces cartilaginosos de BOLD, ya que el nombre de esta clase no aparece en BOLD. Los usuarios deberían utilizar --taxon Elasmobranchii en este caso. Los usuarios también pueden especificar limitar la descarga a un marcador genético específico proporcionando el parámetro --marker . Cuando son de interés varios marcadores genéticos, los nombres de los marcadores deben estar separados por | . Los cuatro marcadores principales de códigos de barras de ADN en BOLD son COI-5P , ITS , matK y rbcL . La entrada para el parámetro --marker distingue entre mayúsculas y minúsculas.
Enfoque recomendado: un bucle for simple para descargar datos de BOLD para múltiples grupos taxonómicos (enfoque recomendado). El siguiente código primero descarga datos de Elasmobranchii, seguido de secuencias asignadas a Mammalia. Los datos descargados se escribirán en el subdirectorio --output crabs_testing y se colocarán en dos archivos separados, indicando qué datos pertenecen a qué grupo taxonómico, es decir, crabs_testing/bold_Elasmobranchii.fasta y crabs_testing/bold_Mammalia.fasta .
for taxon in Elasmobranchii Mammalia; do crabs --download-bold --taxon ${taxon} --output crabs_testing/bold_${taxon}.fasta; done

Opción alternativa: además del bucle for recomendado, se pueden proporcionar varios nombres de taxones a la vez separándolos mediante | .
crabs --download-bold --taxon 'Elasmobranchii|Mammalia' --output crabs_testing/bold_elasmobranchii_mammalia.fasta
--download-embl Las secuencias del EMBL se descargan a través del sitio FTP de ENA. Los archivos EMBL se descargarán primero en formato '.fasta.gz' y se descomprimirán automáticamente una vez que se complete la descarga. Esta base de datos no proporciona tanta flexibilidad con respecto a la descarga selectiva en comparación con BOLD o NCBI. Más bien, los datos del EMBL están estructurados en 15 divisiones fiscales, que se pueden descargar por separado. La división de impuestos a descargar se puede especificar usando el parámetro --taxon . Dado que cada división fiscal se divide en varios archivos, se proporciona un * después del nombre para descargar todos los archivos. Los usuarios también pueden descargar un archivo específico escribiendo el nombre completo del archivo. A continuación se proporciona una lista de las 15 opciones de división fiscal. El directorio de salida y el nombre del archivo se pueden especificar utilizando el parámetro --output .
Lista de divisiones fiscales:
crabs --download-embl --taxon 'mam*' --output crabs_testing/embl_mam.fasta
--download-mitofish CRABS también puede descargar la base de datos MitoFish. Esta base de datos es un único archivo fasta de dos líneas. El directorio de salida y el nombre del archivo se pueden especificar utilizando el parámetro --output .
crabs --download-mitofish --output crabs_testing/mitofish.fasta

--download-ncbi Las secuencias de la base de datos NCBI se descargan a través de las utilidades de programación de Entrez. NCBI permite la descarga de datos de varias bases de datos, que los usuarios pueden especificar con el parámetro --database . Para la mayoría de los usuarios, la base de datos --database nucleotide será la más apropiada para crear una base de datos de referencia local.
Para especificar los datos que se descargarán del NCBI, los usuarios realizan una búsqueda a través del parámetro --query . Elaborar buenas búsquedas NCBI puede resultar complicado. Una buena forma de crear una consulta de búsqueda es utilizar la ventana de búsqueda de la página web del NCBI. Desde este enlace, primero haz una búsqueda inicial y presiona enter. Esto lo llevará a la página de resultados donde podrá refinar aún más su búsqueda. En la captura de pantalla siguiente, hemos refinado aún más la búsqueda limitando la longitud de la secuencia entre 100 y 25 000 pb e incorporando solo secuencias mitocondriales. Los usuarios pueden copiar y pegar el texto en el cuadro "Detalles de búsqueda" del sitio web y proporcionarlo entre comillas en el parámetro --query . Otro beneficio de utilizar la ventana de búsqueda de la página web del NCBI es que la página web mostrará cuántas secuencias coinciden con su consulta de búsqueda, que debe coincidir con la cantidad de secuencias reportadas por CRABS. Esta página web proporciona un breve tutorial adicional sobre el uso de la función de búsqueda en la página web del NCBI que nuestro equipo ha escrito para obtener información adicional.
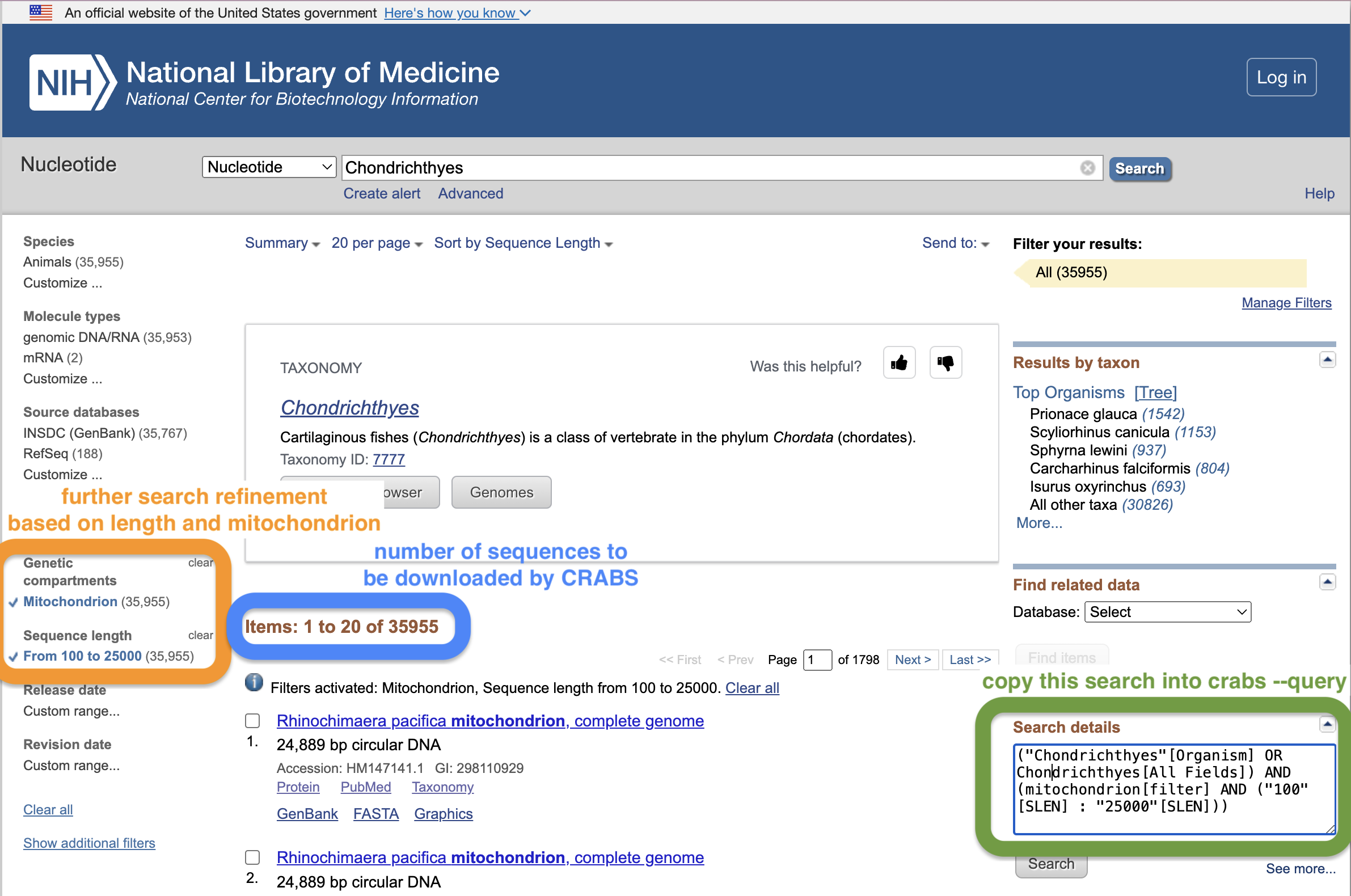
Además de la consulta de búsqueda ( --query ), los usuarios pueden restringir aún más el término de búsqueda descargando datos de secuencia para una lista de especies utilizando el parámetro --species . El parámetro --species toma una cadena de entrada de nombres de especies separados por + o un archivo .txt de entrada con un solo nombre de especie por línea en el documento. El parámetro --batchsize proporciona a los usuarios la opción de descargar secuencias en lotes de N desde el sitio web del NCBI. Este parámetro tiene como valor predeterminado 5.000. No se recomienda aumentar este valor por encima de 5000, ya que lo más probable es que los servidores NCBI desconecten la descarga si se descargan demasiadas secuencias a la vez. El parámetro --email permite a los usuarios especificar su dirección de correo electrónico, que es necesaria para acceder a los servidores de NCBI. Finalmente, el directorio de salida y el nombre del archivo se pueden especificar usando el parámetro --output .
crabs --download-ncbi --query '("Chondrichthyes"[Organism] OR Chondrichthyes[All Fields]) AND (mitochondrion[filter] AND ("100"[SLEN] : "25000"[SLEN]))' --output crabs_testing/ncbi_chondrichthyes.fasta --email [email protected] --database nucleotide

--import Una vez que se descarguen los datos de los repositorios en línea, los archivos deberán importarse a CRABS utilizando la función --import . El formato CRABS constituye una única línea delimitada por tabulaciones por secuencia que contiene toda la información, incluido (i) ID de secuencia, (ii) nombre taxonómico analizado desde la descarga inicial, (iii) número de ID de taxón NCBI, (iv) linaje taxonómico según la taxonomía NCBI. y (v) la secuencia. CRABS intentará obtener el número de acceso del NCBI para cada secuencia como ID de secuencia. Si la secuencia no contiene un número de acceso, es decir, no está depositada en NCBI, CRABS generará ID de secuencia únicas utilizando el siguiente formato: crabs_*[num]*_taxonomic_name . El formato del documento de entrada se especifica mediante el parámetro --import-format y especifica el nombre del repositorio desde el que se descargaron los datos, es decir, BOLD , EMBL , MITOFISH o NCBI . El linaje taxonómico que crea CRABS se basa en la taxonomía NCBI y CRABS requiere que los tres archivos se descarguen usando la función --download-taxonomy , es decir, --names , --nodes y --acc2tax . A partir de la versión v 1.0.0 , CRABS es capaz de resolver sinónimos y nombres no aceptados para incorporar un mayor número de secuencias y diversidad en la base de datos de referencia local. Los rangos taxonómicos que se incluirán en el linaje taxonómico se pueden especificar utilizando los parámetros --ranks . Si bien se puede incluir cualquier rango taxonómico, recomendamos utilizar la siguiente entrada para incluir toda la información necesaria para la mayoría de los clasificadores taxonómicos --ranks 'superkingdom;phylum;class;order;family;genus;species' . El archivo de salida se puede especificar utilizando el parámetro --output y es un archivo .txt simple. En la ventana Terminal, CRABS imprime los resultados del número de secuencias importadas, así como cualquier secuencia para la cual no se pudo generar ningún linaje taxonómico.
crabs --import --import-format bold --input crabs_testing/bold_Elasmobranchii.fasta --names crabs_testing/names.dmp --nodes crabs_testing/nodes.dmp --acc2tax crabs_testing/nucl_gb.accession2taxid --output crabs_testing/crabs_bold.txt --ranks 'superkingdom;phylum;class;order;family;genus;species'

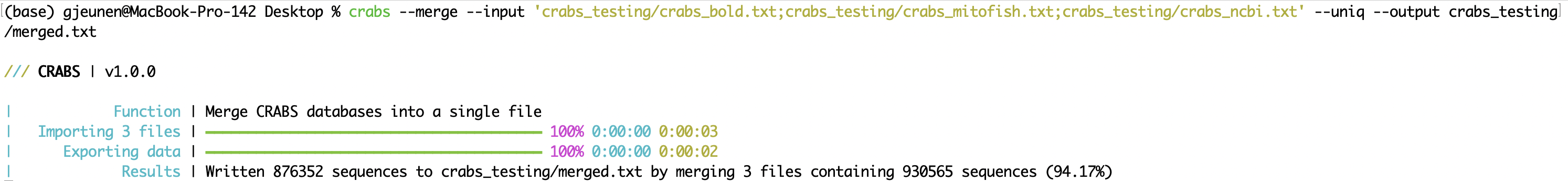
--merge Cuando se descargan datos de secuencia de varios repositorios en línea, los archivos se pueden fusionar en un solo archivo después de importarlos (consulte 5.2.1 --import ) usando la función --merge . Los archivos de entrada para fusionar se pueden ingresar usando el parámetro --input , con los archivos separados por ; . Es posible que una secuencia se haya descargado varias veces cuando se depositó en varios repositorios en línea. El uso del parámetro --uniq conserva solo una versión de cada número de acceso. El archivo de salida se puede especificar utilizando el parámetro --output . En la ventana Terminal, CRABS imprime los resultados de la cantidad de secuencias fusionadas, así como la cantidad de secuencias retenidas cuando se usa el parámetro --uniq .
crabs --merge --input 'crabs_testing/crabs_bold.txt;crabs_testing/crabs_mitofish.txt;crabs_testing/crabs_ncbi.txt' --uniq --output crabs_testing/merged.txt
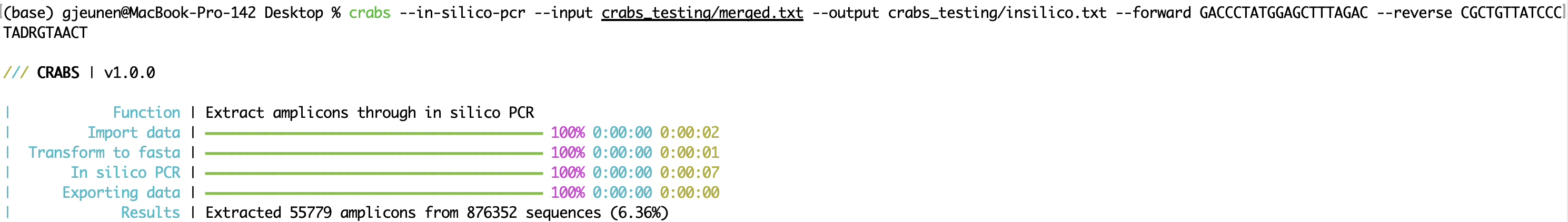
CRABS extrae la región del amplicón del conjunto de cebadores mediante la realización de una PCR in silico (función: --in-silico-pcr ). CRABS utiliza cutadapt v 4.4 para la PCR in silico para aumentar la velocidad de ejecución del código Python tradicional. Los nombres de los archivos de entrada y salida se pueden especificar utilizando los parámetros ' --input ' y ' --output ', respectivamente. Tanto el cebador directo como el inverso deben proporcionarse en la dirección 5'-3' utilizando los parámetros ' --forward ' y ' --reverse ', respectivamente. CANGREJOS complementará de forma inversa la imprimación inversa. Desde la versión v 1.0.0 , CRABS es capaz de retener códigos de barras en ambas direcciones mediante un único análisis de PCR in silico . Por lo tanto, no se realiza ningún paso de complementación inversa ni se vuelve a ejecutar la PCR in silico , lo que aumenta significativamente la velocidad de ejecución. Para conservar secuencias para las que no se pudieron encontrar regiones de unión de cebadores, se puede especificar un archivo de salida para el parámetro --untrimmed . El número máximo permitido de discrepancias encontradas en las regiones de unión del cebador se puede especificar utilizando el parámetro --mismatch , con una configuración predeterminada de 4. Finalmente, el análisis de PCR in silico puede ser multiproceso en CRABS. De forma predeterminada, se utiliza la cantidad máxima de subprocesos, pero los usuarios pueden especificar la cantidad de subprocesos a usar con el parámetro --threads .
crabs --in-silico-pcr --input crabs_testing/merged.txt --output crabs_testing/insilico.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT
Es una práctica común eliminar las regiones de unión a cebadores de las secuencias de referencia cuando se depositan en una base de datos en línea. Por lo tanto, cuando la secuencia de referencia se generó utilizando el mismo cebador directo y/o inverso que se busca en la función --in-silico-pcr , la función --in-silico-pcr no habrá podido recuperar la región amplicón del secuencia de referencia. Para tener en cuenta esta posibilidad, CRABS tiene la opción de ejecutar una alineación global por pares, implementada con VSEARCH v 2.16.0 , para extraer regiones de amplicones para las cuales la secuencia de referencia no contiene las regiones de unión de cebadores directa e inversa completas. Para lograr esto, la función --pairwise-global-alignment toma el archivo de base de datos descargado originalmente usando el parámetro --input . La base de datos en la que se buscará es el archivo de salida de --in-silico-pcr y se puede especificar usando el parámetro --amplicons . El archivo de salida se puede especificar utilizando el parámetro --output . Las secuencias de cebadores, que solo se utilizan para calcular la longitud del par de bases, se pueden configurar con los parámetros --forward y --reverse . Como la función --pairwise-global-alignment puede tardar mucho en ejecutarse en bases de datos grandes, la longitud de la secuencia se puede restringir para acelerar el proceso utilizando el parámetro --size-select . El porcentaje mínimo de identidad y cobertura de consulta se pueden especificar utilizando los parámetros --percent-identity y --coverage , respectivamente. --percent-identity debe proporcionarse como un valor porcentual entre 0 y 1 (por ejemplo, 95 % = 0,95), mientras que --coverage debe proporcionarse como un valor porcentual entre 0 y 100 (por ejemplo, 95 % = 95). De forma predeterminada, la función --pairwise-global-alignment está restringida a retener secuencias donde las secuencias del cebador no están completamente presentes en la secuencia de referencia (la alineación comienza o termina dentro de la longitud del cebador directo o inverso). Cuando se proporciona el parámetro --all-start-positions , se incluirán aciertos positivos cuando la alineación se encuentre fuera del rango de las regiones de unión del cebador (omitido por la función --in-silico-pcr debido a demasiadas discrepancias en el región de unión del cebador). No recomendamos usar --all-start-positions , ya que es muy poco probable que se amplifique un código de barras usando el conjunto de cebadores especificado de la función --in-silico-pcr cuando hay más de 4 discrepancias presentes en el cebador- regiones de unión.
crabs --pairwise-global-alignment --input crabs_testing/merged.txt --amplicons crabs_testing/insilico.txt --output crabs_testing/aligned.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT --size-select 10000 --percent-identity 0.95 --coverage 0.95
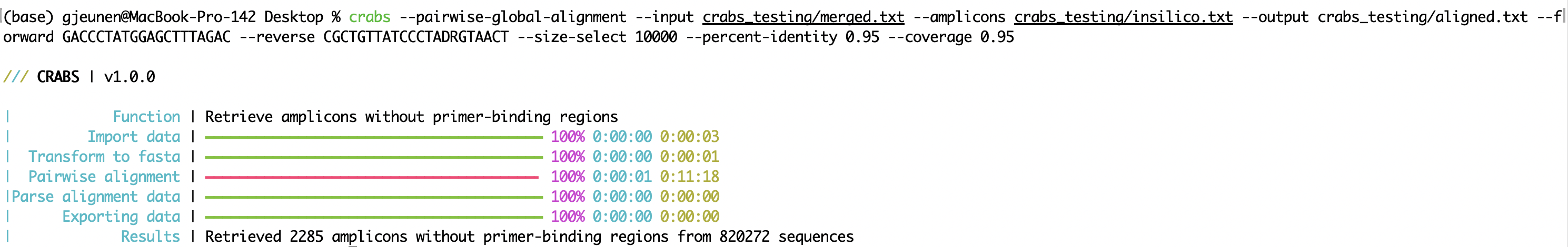
--pairwise-global-alignment La función --pairwise-global-alignment puede tardar una cantidad considerable de tiempo en ejecutarse cuando CRABS procesa archivos de secuencia grandes, aunque se admita multiproceso. Desde la actualización a CRABS v 1.0.0 , existe una estructura de archivos idéntica desde --import hasta --export , lo que permite que las funciones se ejecuten en cualquier orden. Si bien todavía recomendamos seguir el orden del flujo de trabajo de CRABS, la función --pairwise-global-alignment se puede acelerar significativamente al ejecutar las funciones --dereplicate y --filter antes de la función --in-silico-pcr . Al ejecutar estos pasos de curación antes de --in-silico-pcr , la cantidad de secuencias que CRABS debe procesar para la función --pairwise-global-alignment se reducirá significativamente.
NOTA 1 : al ejecutar la función --filter antes de --in-silico-pcr , asegúrese de omitir cualquier parámetro que afecte directamente a la secuencia, ya que --filter basará esto en la secuencia completa y no en el amplicón extraído. . Por lo tanto, omita los siguientes parámetros: --minimum-length , --maximum-length , --maximum-n .
NOTA 2 : al ejecutar las funciones --dereplicate y --filter antes de --in-silico-pcr , sería recomendable ejecutar ambas funciones nuevamente después de --pairwise-global-alignment , ya que la base de datos podría curarse aún más ahora que se extraigan los amplicones.
Una vez que las funciones --in-silico-pcr y --pairwise-global-alignment hayan extraído todos los códigos de barras potenciales para el conjunto de cebadores, la base de datos de referencia local puede someterse a una mayor curación y subconjunto dentro de CRABS usando varias funciones, incluida --dereplicate , --filter y --subset .
--dereplicate El primer método de curación es eliminar la replicación de la base de datos de referencia local utilizando la función --dereplicate . Es posible que, para ciertos taxones, en este momento existan múltiples códigos de barras idénticos en la base de datos de referencia local. Esto puede ocurrir cuando diferentes grupos de investigación han depositado secuencias idénticas o si la variación intraespecífica entre secuencias de un taxón no está contenida en el código de barras extraído. Es mejor eliminar estos códigos de barras de referencia idénticos para acelerar la asignación de taxonomías, así como para mejorar los resultados de la asignación de taxonomías (especialmente para clasificadores taxonómicos que proporcionan un número limitado de resultados, es decir, BLAST).
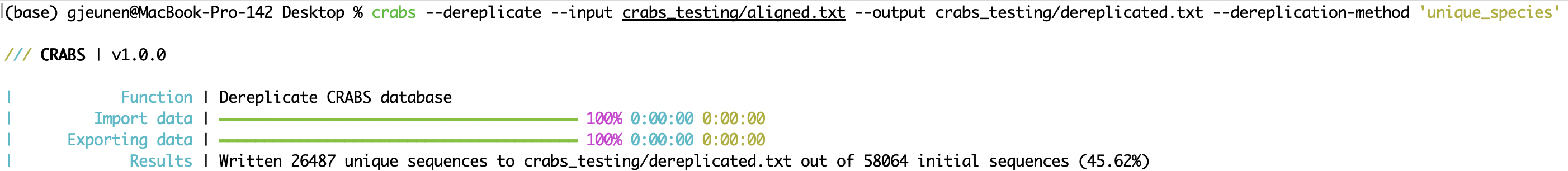
Los archivos de entrada y salida se pueden especificar utilizando los parámetros --input y --output , respectivamente. CRABS ofrece tres métodos de desreplicación, que se pueden especificar utilizando el parámetro --dereplication-method , que incluye:
crabs --dereplicate --input crabs_testing/aligned.txt --output crabs_testing/dereplicated.txt --dereplication-method 'unique_species'
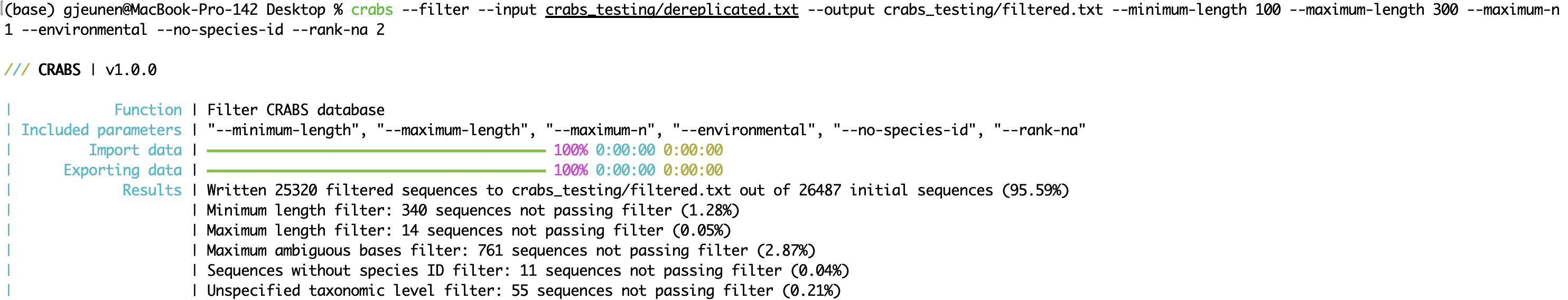
--filter El segundo método de curación es filtrar la base de datos de referencia local utilizando varios parámetros utilizando la función --filter . Los archivos de entrada y salida se pueden especificar utilizando los parámetros --input y --output , respectivamente. Desde la versión v 1.0.0 . CRABS incorpora el filtrado basado en seis parámetros, entre ellos:
--minimum-length : longitud mínima de secuencia para que un amplicón se conserve en la base de datos;--maximum-length : longitud máxima de secuencia para que un amplicón se conserve en la base de datos;--maximum-n : descarta amplicones con N o bases más ambiguas ( N );--environmental : descarta secuencias ambientales de la base de datos;--no-species-id : descarta secuencias para las que no hay ningún nombre de especie disponible;--rank-na : descarta secuencias con N o más niveles taxonómicos no especificados. crabs --filter --input crabs_testing/dereplicated.txt --output crabs_testing/filtered.txt --minimum-length 100 --maximum-length 300 --maximum-n 1 --environmental --no-species-id --rank-na 2
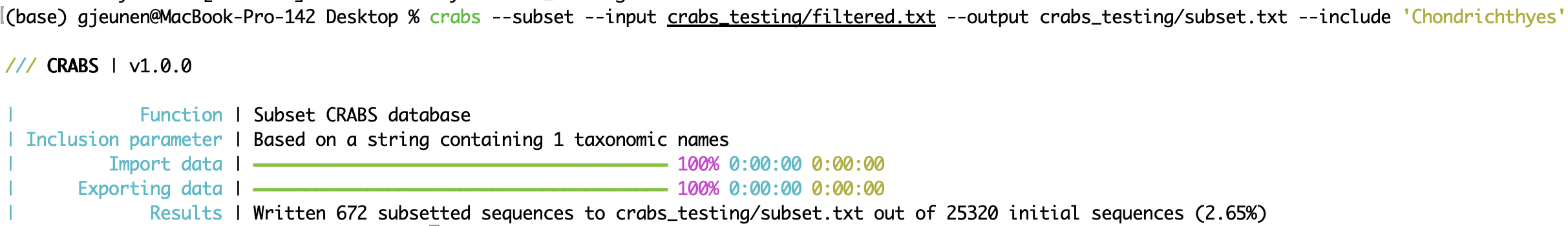
--subset El tercer y último método de curación incorporado en CRABS es subconjunto de la base de datos de referencia local para incluir (parámetro: --include ) o excluir (parámetro: --exclude ) taxones específicos usando la función --subset . Esta función permite eliminar códigos de barras de referencia de grupos taxonómicos que no son de interés para la pregunta de investigación. Estos grupos taxonómicos podrían haberse incorporado a la base de datos de referencia local debido a una posible amplificación inespecífica del conjunto de cebadores. Otro caso de uso de --subset es eliminar secuencias erróneas conocidas.
Para los clasificadores taxonómicos basados en aprendizaje automático (IDTAXA) o distancia k-mer (SINTAX), puede ser beneficioso subconjunto de la base de datos de referencia incluyendo solo taxones que se sabe que existen en la región donde se tomaron las muestras y excluyendo especies estrechamente relacionadas que se sabe que no existen. que ocurra en la región para aumentar la resolución taxonómica obtenida de estos clasificadores y obtener mejores resultados de asignación de taxonomía.
Los archivos de entrada y salida se pueden especificar utilizando los parámetros --input y --output , respectivamente. Los parámetros --include y --exclude pueden incluir una lista de taxones separados por ; o un archivo .txt que contenga un único nombre de taxón por línea.
crabs --subset --input crabs_testing/filtered.txt --output crabs_testing/subset.txt --include 'Chondrichthyes'
Una vez finalizada la base de datos de referencia, se puede exportar a varios formatos para adaptarse a las especificaciones requeridas por la mayoría de las herramientas de software que asignan taxonomía a datos metagenómicos. Los archivos de entrada y salida se pueden especificar utilizando los parámetros --input y --output , respectivamente. A partir de la versión v 1.0.0 , CRABS incorpora el formateo de la base de datos de referencia para seis clasificadores diferentes (parámetro: --export-format ), incluyendo:
--export-format 'sintax' : El clasificador SINTAX está incorporado en USEARCH y VSEARCH;--export-format 'rdp' : el clasificador RDP es un programa independiente ampliamente utilizado en estudios de microbioma;--export-format 'qiime-fasta' y --export-format 'qiime-text' : se puede utilizar para asignar una identificación taxonómica en QIIME y QIIME2;--export-format 'dada2-species' y --export-format 'dada2-taxonomy' : se puede utilizar para asignar una identificación taxonómica en DADA2;--export-format 'idt-fasta' y --export-format 'idt-text' : el clasificador IDTAXA es un algoritmo de aprendizaje automático incorporado en el paquete DECIPHER R;--export-format 'blast-notax' : Crea una base de datos de referencia de BLAST local para BLASTN y Megablast donde el resultado no proporciona una ID taxonómica, pero enumera el número de acceso;--export-format 'blast-tax' : Crea una base de datos de referencia de BLAST local para BLASTN y MEGABLAST, donde el resultado proporciona la identificación taxonómica y el número de acceso. crabs --export --input crabs_testing/subset.txt --output crabs_testing/BLAST_TAX_CHONDRICHTHYES --export-format 'blast-tax'

Al exportar la base de datos de referencia local a un formato único (excepto los clasificadores donde la base de datos de referencia se divide en múltiples archivos, es decir, Qiime, DADA2, IDTAXA) será suficiente para la mayoría de los usuarios, se puede escribir un bucle simple para exportar el local Base de datos de referencia a múltiples formatos Si los usuarios desean comparar resultados entre diferentes clasificadores taxonómicos. A continuación se proporciona un ejemplo para exportar la base de datos de referencia local en formatos Sintax, RDP e IDTAXA.
for format in sintax.fasta rdp.fasta idt-fasta.fasta idt-text.txt; do crabs --export --input crabs_testing/subset.txt --output crabs_testing/chondrichthyes_${format} --export-format ${format%%.*}; done

Una vez que se finaliza la base de datos de referencia, los cangrejos pueden ejecutar cinco funciones de postprocesamiento para explorar y proporcionar una descripción resumida de la base de datos de referencia local, que incluye (i) --diversity-figure , (ii) --amplicon-length-figure ((( iii) --phylogenetic-tree , (iv) --amplification-efficiency-figure y (v) --completeness-table .
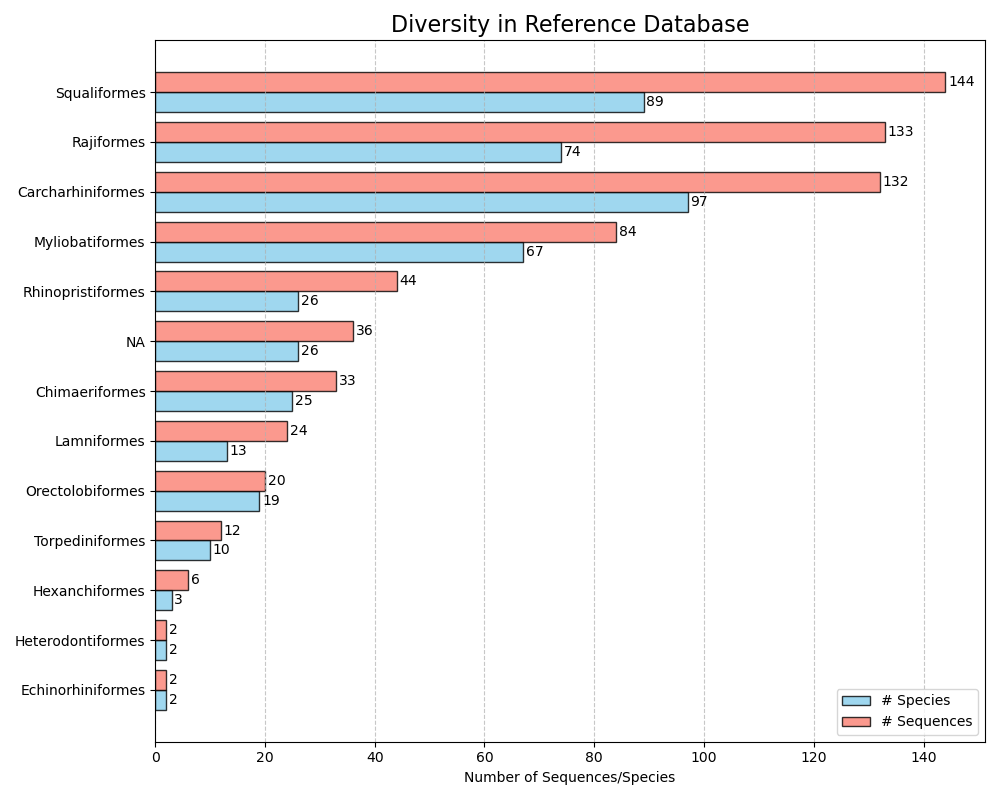
--diversity-figure La función --diversity-figure produce una gráfica de barra horizontal con número de especies (en azul) y número de secuencias (en naranja) por para cada grupo taxonómico en la base de datos de referencia. El usuario puede especificar el rango taxonómico para dividir la base de datos de referencia con el parámetro --tax-level . El nivel de impuestos es el número del rango en el que apareció durante la función --import . Por ejemplo, si --ranks 'superkingdom;phylum;class;order;family;genus;species' se usó durante-división --import basada en superkingdom requeriría --tax-level 1 , phylum = --tax-level 2 , class = --tax-level 3 , etc. El archivo de entrada en formato de cangrejos se puede especificar utilizando el parámetro --input . La figura, en formato .png, se escribirá en el archivo de salida, que se puede especificar utilizando el parámetro --output .
crabs --diversity-figure --input crabs_testing/subset.txt --output crabs_testing/diversity-figure.png --tax-level 4
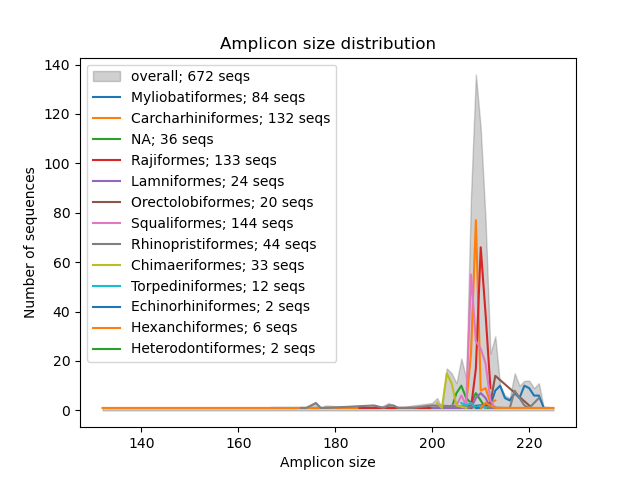
--amplicon-length-figure La función --amplicon-length-figure produce un gráfico de línea que muestra el rango de la longitud del amplicón. El rango general en la longitud del amplicón en todas las secuencias en la base de datos de referencia se muestra en un color gris sombreado, mientras que los resultados se dividen por grupo taxonómico (parámetro: --tax-level ) se superponen por líneas de colores. Además, la leyenda muestra el número de secuencias asignadas a cada uno de los grupos taxonómicos y el número total de secuencias en la base de datos de referencia. El archivo de entrada en formato de cangrejos se puede especificar utilizando el parámetro --input . La figura, en formato .png, se escribirá en el archivo de salida, que se puede especificar utilizando el parámetro --output .
crabs --amplicon-length-figure --input crabs_testing/subset.txt --output crabs_testing/amplicon-length-figure.png --tax-level 4
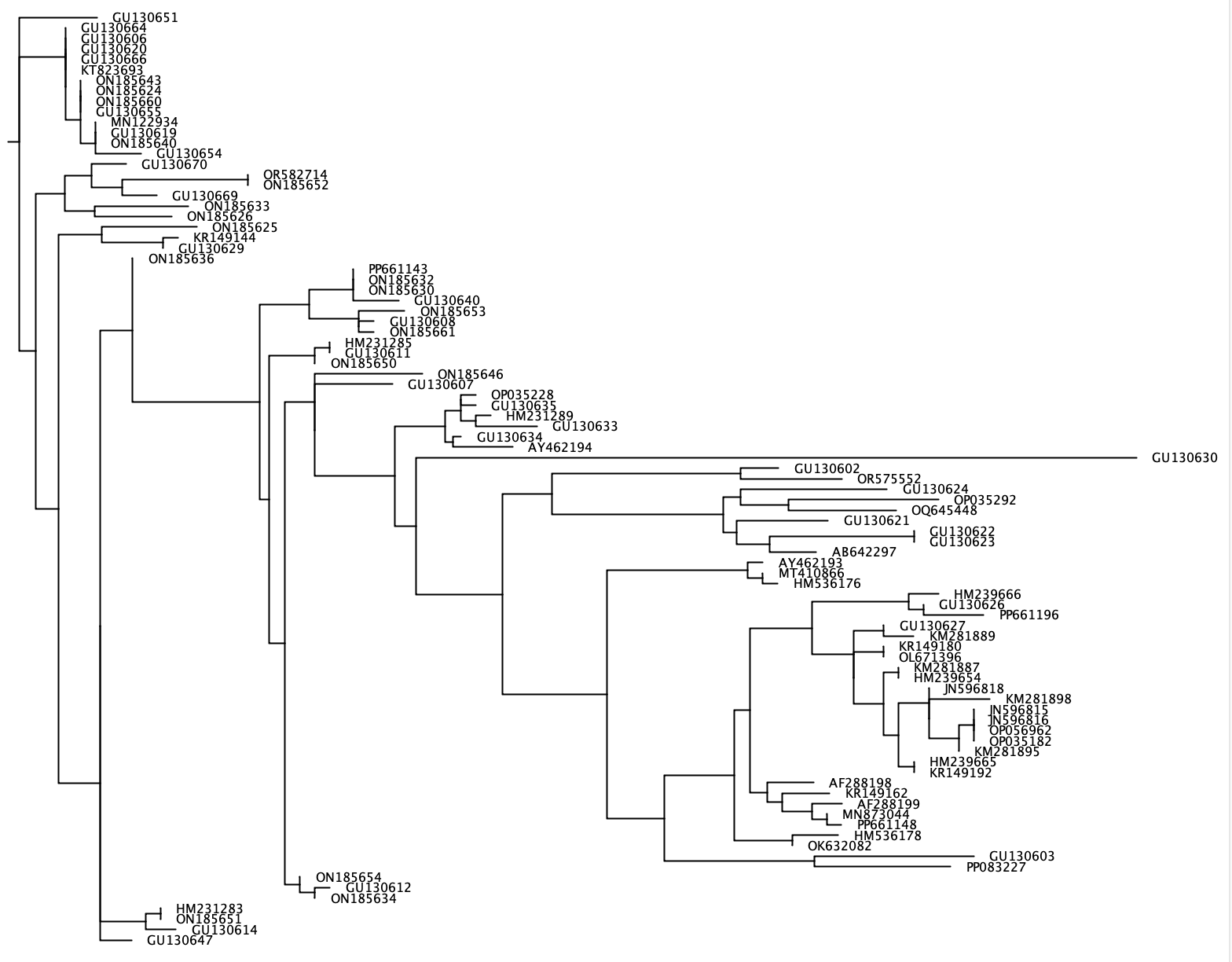
--phylogenetic-tree La función --phylogenetic-tree generará un árbol filogenético para una lista de especies de interés. Esta lista de especies de interés se puede importar utilizando el parámetro --species y consiste en una cadena de entrada separada por + o un archivo .txt con un solo nombre de especie en cada línea. Para cada especie de interés, las secuencias se extraerán de la base de datos de referencia que comparten un rango taxonómico definido por el usuario (parámetro: --tax-level ) con las especies de interés. Los cangrejos generarán una alineación de todas las secuencias extraídas utilizando Clustalw2 V 2.1 y generarán un árbol filogenético que une al vecino usando FastTree. Este árbol filogenético en formato Newick se escribirá en el archivo de salida utilizando el parámetro --output y se puede visualizar en programas de software como FigTree o Geneious. Dado que se generará un árbol filogenético separado para cada especie de interés, --output toma un nombre de archivo genérico, mientras que el archivo de salida exacto contendrá este nombre genérico seguido de '_species_name.tree'.
crabs --phylogenetic-tree --input crabs_testing/subset.txt --output crabs_testing/phylo --tax-level 4 --species 'Carcharodon carcharias+Squalus acanthias'
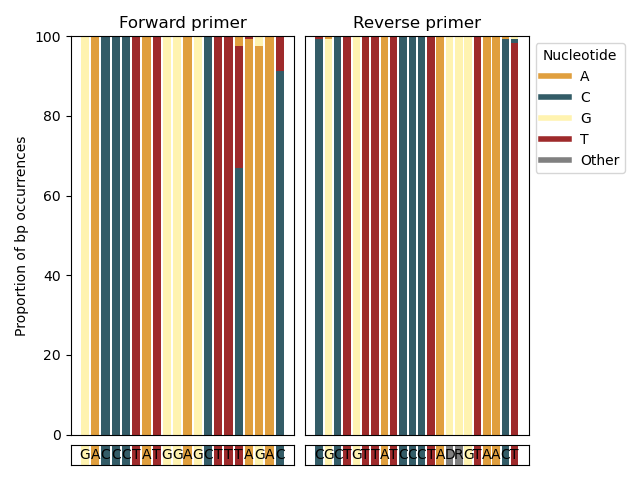
--amplification-efficiency-figure La función --amplification-efficiency-figure producirá un gráfico de barras, que muestra la proporción de la ocurrencia de pares de bases en las regiones de unión a cebadores para un grupo taxonómico especificado por el usuario, visualizando así los lugares en las regiones de unión a los imprimadores hacia adelante e inversa donde no coinciden. podría estar ocurriendo en el grupo taxonómico de intereses, lo que puede influir en la eficiencia de la amplificación. La función --amplification-efficiency-figure toma una base de datos de referencia de cangrejos final como entrada utilizando el parámetro --amplicons . Para encontrar la información en las regiones de unión a cebadores para cada secuencia en el archivo de entrada, las secuencias inicialmente descargadas después de la importación deben proporcionarse utilizando el parámetro --input . Las secuencias de cebador directo e inversa (en la dirección 5 ' -3') se proporcionan utilizando los parámetros --forward y --reverse . El nombre del grupo taxonómico de intereses se puede proporcionar utilizando el parámetro --tax-group y se puede establecer en cualquier nivel taxonómico que se incorpore en el archivo de entrada. Finalmente, la figura en formato .png se escribirá en el archivo de salida especificado por el parámetro --output .
crabs --amplification-efficiency-figure --input crabs_testing/merged.txt --amplicons crabs_testing/subset.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT --output crabs_testing/amplification-efficiency.png --tax-group Carcharhiniformes
--completeness-table La función --completeness-table generará una tabla delimitada de pestañas (parámetro: --output ) con información sobre una lista de especies de interés. Esta lista de especies de interés se puede importar utilizando el parámetro --species y consiste en una cadena de entrada separada por + o un archivo .txt con un solo nombre de especie en cada línea. Se generará un linaje taxonómico para cada especie de interés utilizando los archivos ' Names.dmp ' y ' nodos.dmp ' descargados utilizando la función --download-taxonomy utilizando los parámetros --names y --nodes , respectivamente. La tabla de salida tendrá 10 columnas que proporcionan la siguiente información:
crabs --completeness-table --input crabs_testing/subset.txt --output crabs_testing/completeness.txt --names crabs_testing/names.dmp --nodes crabs_testing/nodes.dmp --species 'Carcharodon carcharias+Squalus acanthias'
crabs --version v 1.0.6 : corrección de errores -> Analización mejorada de encabezados en negrita durante --import .crabs --version v 1.0.5 : corrección de errores -> Se agregó una restricción de longitud a la identificación SEQ al construir bases de datos de explosión, según sea necesario para el software BLAST+.crabs --version v 1.0.4 : Información agregada-> proporcionó información correcta sobre la entrada de valor para --pairwise-global-alignment --coverage --percent-identity .crabs --version v 1.0.3 : corrección de errores -> Verificación de la respuesta del servidor NCBI 3 veces antes de abortar el análisis.crabs --version v 1.0.2 : corrección de errores -> capaz de informar cuándo se devuelven 0 secuencias después del análisis.crabs --version v 1.0.1 : corrección de errores -> consulta de construcción NCBI exitosa utilizando el parámetro --species .

