nucleotide transformer
1.0.0
Bienvenido a este repositorio InstaDeep Github, donde se presentan:
Estamos encantados de abrir el código fuente de estos trabajos y brindar a la comunidad acceso al código y a los pesos previamente entrenados para estos nueve modelos de lenguaje genómico y dos modelos de segmentación. Los modelos del proyecto nucleotide transformer se desarrollaron en colaboración con Nvidia y TUM, y los modelos se entrenaron en nodos DGX A100 en Cambridge-1. El modelo del proyecto nucleotide transformer Agro se desarrolló en colaboración con Google y se entrenó en aceleradores TPU-v4.
En general, nuestro trabajo proporciona conocimientos novedosos relacionados con el entrenamiento previo y la aplicación de modelos fundamentales del lenguaje, así como el entrenamiento de modelos que los utilizan como codificador principal, para la genómica con amplias oportunidades de sus aplicaciones en el campo.
En este repositorio encontrará lo siguiente:
En comparación con otros enfoques, nuestros modelos no solo integran información de genomas de referencia únicos, sino que aprovechan secuencias de ADN de más de 3200 genomas humanos diversos, así como 850 genomas de una amplia gama de especies, incluidos organismos modelo y no modelo. A través de una evaluación sólida y extensa, demostramos que estos grandes modelos proporcionan una predicción de fenotipo molecular extremadamente precisa en comparación con los métodos existentes.
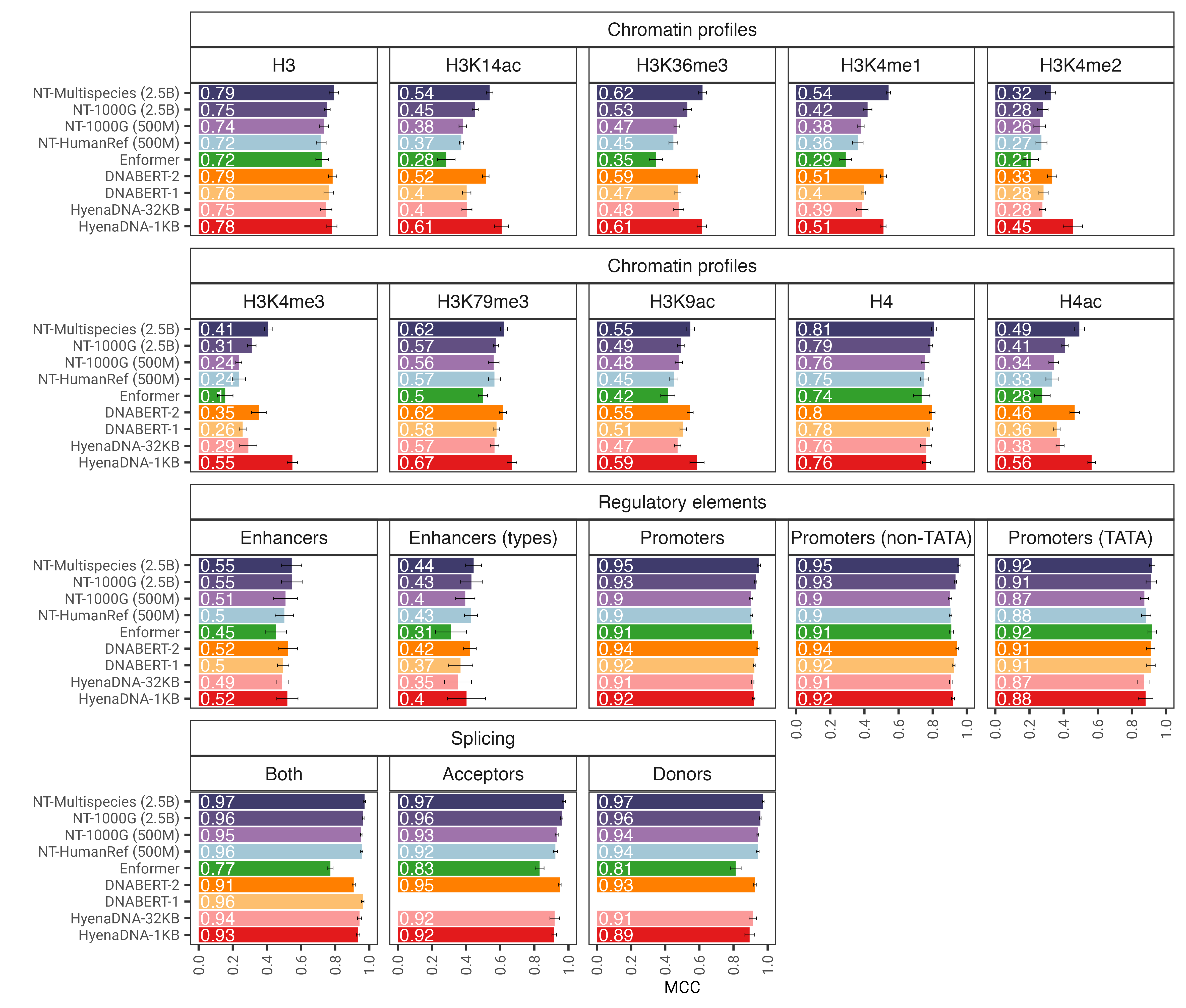
Fig. 1: El modelo nucleotide transformer predice con precisión diversas tareas genómicas después del ajuste fino. Mostramos los resultados de rendimiento en las tareas posteriores para modelos de transformadores ajustados. Las barras de error representan 2 DE derivadas de una validación cruzada de 10 veces.
En este trabajo presentamos un novedoso modelo fundamental de lenguaje grande entrenado en genomas de referencia de 48 especies de plantas con un enfoque predominante en especies de cultivos. Evaluamos el desempeño de AgroNT en varias tareas de predicción que van desde características regulatorias, procesamiento de ARN y expresión genética, y demostramos que AgroNT puede obtener un desempeño de vanguardia.
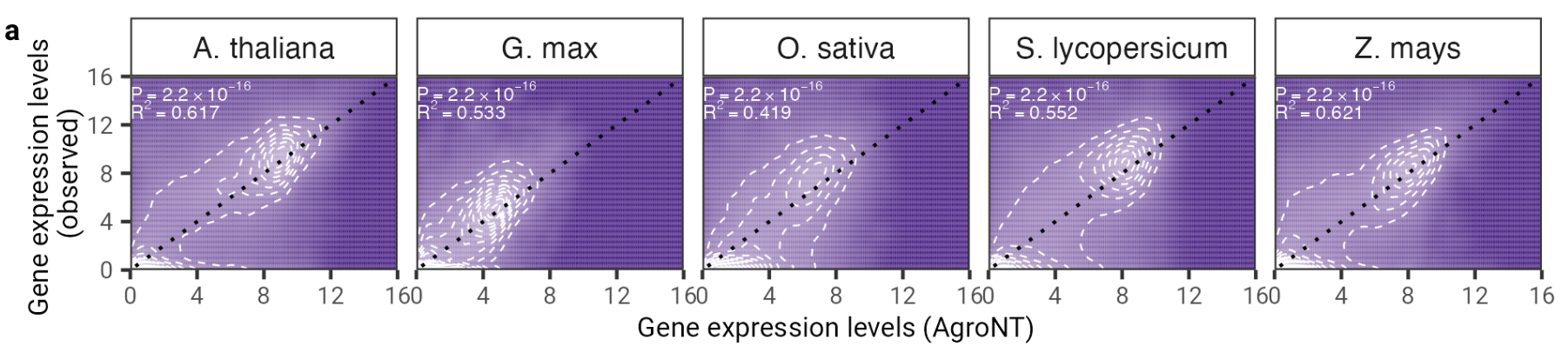
Fig. 2: AgroNT proporciona predicción de la expresión genética en diferentes especies de plantas. La predicción de la expresión genética en genes reservados en todos los tejidos se correlaciona con los niveles de expresión genética observados. Se muestran el coeficiente de determinación (R 2 ) de un modelo lineal y los valores P asociados entre los valores previstos y observados.
Para utilizar el código y los modelos previamente entrenados, simplemente:
pip install . .Luego puedes descargar y hacer la inferencia con cualquiera de nuestros nueve modelos en solo unas pocas líneas de código:
import haiku as hk
import jax
import jax . numpy as jnp
from nucleotide_transformer . pretrained import get_pretrained_model
# Get pretrained model
parameters , forward_fn , tokenizer , config = get_pretrained_model (
model_name = "500M_human_ref" ,
embeddings_layers_to_save = ( 20 ,),
max_positions = 32 ,
)
forward_fn = hk . transform ( forward_fn )
# Get data and tokenize it
sequences = [ "ATTCCGATTCCGATTCCG" , "ATTTCTCTCTCTCTCTGAGATCGATCGATCGAT" ]
tokens_ids = [ b [ 1 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens_str = [ b [ 0 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens = jnp . asarray ( tokens_ids , dtype = jnp . int32 )
# Initialize random key
random_key = jax . random . PRNGKey ( 0 )
# Infer
outs = forward_fn . apply ( parameters , random_key , tokens )
# Get embeddings at layer 20
print ( outs [ "embeddings_20" ]. shape )Los nombres de modelos admitidos son:
También puede ejecutar nuestros modelos y encontrar más códigos de ejemplo en Google Colab.
¡El código se ejecuta tanto en GPU como en TPU gracias a Jax!
Nuestra segunda versión de los modelos nucleotide transformer v2 incluye una serie de cambios arquitectónicos que demostraron ser más eficientes: en lugar de utilizar incrustaciones posicionales aprendidas, utilizamos incrustaciones rotativas que se utilizan en cada capa de atención y unidades lineales cerradas con activaciones de movimiento sin sesgo. Estos modelos mejorados también aceptan secuencias de hasta 2048 tokens, lo que conduce a una ventana de contexto más larga de 12 kbp. Inspirándonos en las leyes de escalamiento de Chinchilla, también entrenamos nuestros modelos NT-v2 en nuestro conjunto de datos de múltiples especies para una mayor duración (tokens de 300 mil millones para los modelos 50M y 100M; tokens de 1T para los modelos 250M y 500M) en comparación con los modelos v1 (tokens de 300 mil millones). para los cuatro modelos).
Las capas del transformador están indexadas en 1, lo que significa que llamar get_pretrained_model con los argumentos model_name="500M_human_ref" y embeddings_layers_to_save=(1, 20,) dará como resultado la extracción de incrustaciones después de la primera y la vigésima capa del transformador. Para los transformadores que utilizan el cabezal Roberta LM, es una práctica común extraer las incrustaciones finales después de la norma de la primera capa del cabezal LM en lugar de después del último bloque del transformador. Por lo tanto, si se llama get_pretrained_model con los siguientes argumentos embeddings_layers_to_save=(24,) , las incrustaciones no se extraerán después de la capa final del transformador sino después de la norma de la primera capa del cabezal LM.
Los modelos SegmentNT aprovechan un nucleotide transformer (NT) del cual retiramos el cabezal del modelo de lenguaje y lo reemplazamos por un cabezal de segmentación U-Net unidimensional para predecir la ubicación de varios tipos de elementos genómicos en una secuencia con una resolución de un solo nucleótido. Presentamos dos variantes de modelo diferentes en 14 clases diferentes de elementos genómicos humanos en secuencias de entrada de hasta 30 kb. Estos incluyen genes (genes codificadores de proteínas, lncRNA, 5'UTR, 3'UTR, exón, intrón, aceptor de empalme y sitios donantes) y reguladores (señal poliA, promotores y potenciadores invariantes de tejido y específicos de tejido, y receptores unidos a CTCF). sitios) elementos. SegmentNT logra un rendimiento superior sobre la arquitectura de segmentación U-Net de última generación, beneficiándose de los pesos previamente entrenados de NT y demuestra una generalización cero hasta 50 kbp.
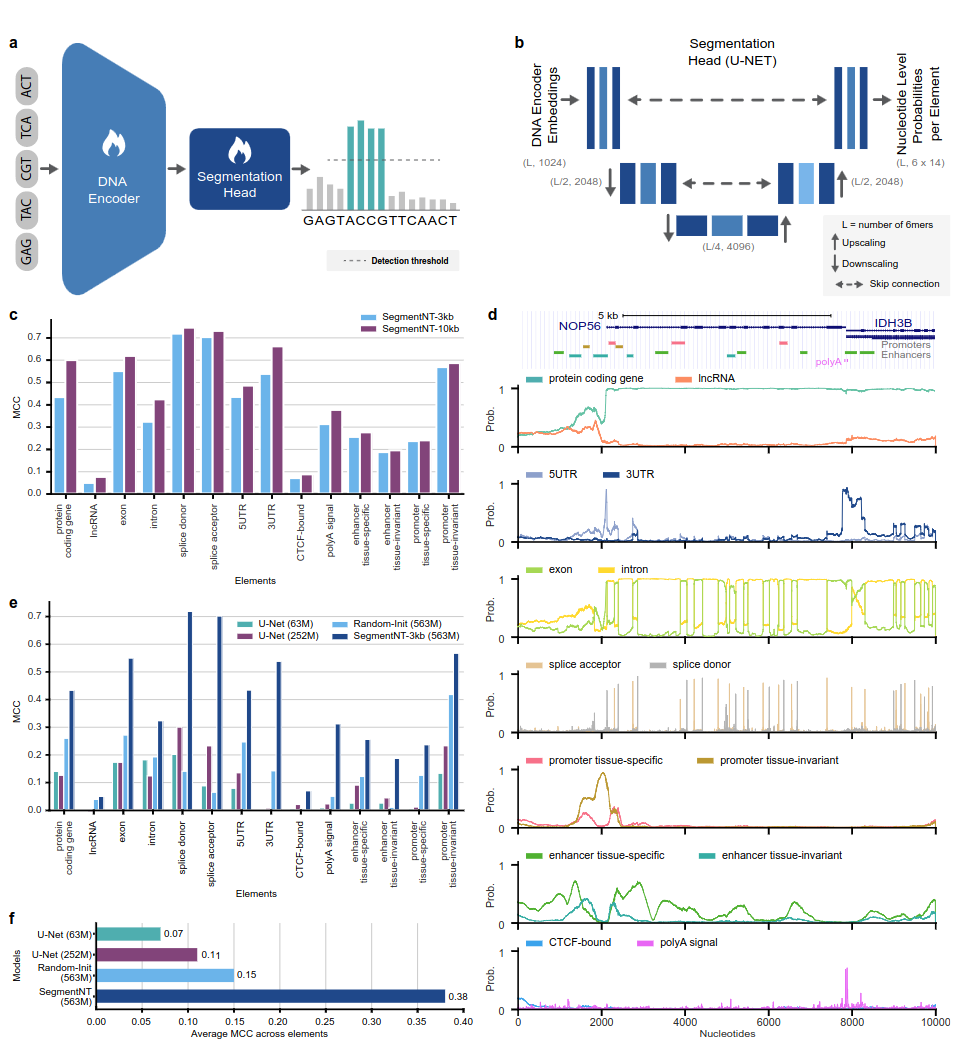
Fig. 1: SegmentNT localiza elementos genómicos con resolución de nucleótidos.
Para utilizar el código y los modelos previamente entrenados, simplemente:
pip install . .Luego podrás descargar e inferir una secuencia con cualquiera de nuestros modelos en solo unas pocas líneas de códigos:
rescaling factor se establece en el utilizado durante el entrenamiento. En caso de que necesite inferir secuencias entre 30 kbp y 50 kbp, asegúrese de pasar el argumento rescaling_factor en la función get_pretrained_segment_nt_model con el valor rescaling_factor = max_num_nucleotides / max_num_tokens_nt donde num_dna_tokens_inference es el número de tokens en la inferencia (es decir, 6669 para una secuencia de 40008 bases pares) y max_num_tokens_nt es el número máximo de tokens en los que se entrenó el transformador de nucleótidos de la columna vertebral, es decir, 2048 .
? El cuaderno de examples/inference_segment_nt.ipynb muestra cómo inferir una secuencia de 50 kb y trazar las probabilidades para reproducir la figura 3 del artículo.
? Los modelos SegmentNT no manejan ninguna "N" en la secuencia de entrada porque cada nucleótido debe tokenizarse como 6 meros, lo que no puede ser el caso cuando se utilizan secuencias que contienen uno o varios pares de bases "N".
import haiku as hk
import jax
import jax . numpy as jnp
from nucleotide_transformer . pretrained import get_pretrained_segment_nt_model
# Initialize CPU as default JAX device. This makes the code robust to memory leakage on
# the devices.
jax . config . update ( "jax_platform_name" , "cpu" )
backend = "cpu"
devices = jax . devices ( backend )
num_devices = len ( devices )
print ( f"Devices found: { devices } " )
# The number of DNA tokens (excluding the CLS token prepended) needs to be dividible by
# 2 to the power of the number of downsampling block, i.e 4.
max_num_nucleotides = 8
assert max_num_nucleotides % 4 == 0 , (
"The number of DNA tokens (excluding the CLS token prepended) needs to be dividible by"
"2 to the power of the number of downsampling block, i.e 4." )
parameters , forward_fn , tokenizer , config = get_pretrained_segment_nt_model (
model_name = "segment_nt" ,
embeddings_layers_to_save = ( 29 ,),
attention_maps_to_save = (( 1 , 4 ), ( 7 , 10 )),
max_positions = max_num_nucleotides + 1 ,
)
forward_fn = hk . transform ( forward_fn )
apply_fn = jax . pmap ( forward_fn . apply , devices = devices , donate_argnums = ( 0 ,))
# Get data and tokenize it
sequences = [ "ATTCCGATTCCGATTCCAACGGATTATTCCGATTAACCGATTCCAATT" , "ATTTCTCTCTCTCTCTGAGATCGATGATTTCTCTCTCATCGAACTATG" ]
tokens_ids = [ b [ 1 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens_str = [ b [ 0 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens = jnp . asarray ( tokens_ids , dtype = jnp . int32 )
random_key = jax . random . PRNGKey ( seed = 0 )
keys = jax . device_put_replicated ( random_key , devices = devices )
parameters = jax . device_put_replicated ( parameters , devices = devices )
tokens = jax . device_put_replicated ( tokens , devices = devices )
# Infer on the sequence
outs = apply_fn ( parameters , keys , tokens )
# Obtain the logits over the genomic features
logits = outs [ "logits" ]
# Transform them in probabilities
probabilities = jnp . asarray ( jax . nn . softmax ( logits , axis = - 1 ))[..., - 1 ]
print ( f"Probabilities shape: { probabilities . shape } " )
print ( f"Features inferred: { config . features } " )
# Get probabilities associated with intron
idx_intron = config . features . index ( "intron" )
probabilities_intron = probabilities [..., idx_intron ]
print ( f"Intron probabilities shape: { probabilities_intron . shape } " )Los nombres de modelos admitidos son:
¡El código se ejecuta tanto en GPU como en TPU gracias a Jax!
Los modelos se entrenan en secuencias de hasta 1000 tokens, incluido el token <CLS> que se antepone automáticamente al comienzo de la secuencia. El tokenizador comienza a tokenizar de izquierda a derecha agrupando las letras "A", "C", "G" y "T" en 6 unidades. La letra "N" se elige para no agruparse dentro de los k-meros, por lo tanto, siempre que el tokenizador encuentre una "N", o si el número de nucleótidos en la secuencia no es múltiplo de 6, tokenizará los nucleótidos sin agruparlos. a ellos. A continuación se dan ejemplos:
dna_sequence_1 = "ACGTGTACGTGCACGGACGACTAGTCAGCA"
tokenized_dna_sequence_1 = [ < CLS > , < ACGTGT > , < ACGTGC > , < ACGGAC > , < GACTAG > , < TCAGCA > ]
dna_sequence_2 = "ACGTGTACNTGCACGGANCGACTAGTCTGA"
tokenized_dna_sequence_2 = [ < CLS > , < ACGTGT > , < A > , < C > , < N > , < TGCACG > , < G > , < A > , < N > , < CGACTA > , < GTCTGA > ]Por lo tanto, todos los transformadores v1 y v2 pueden tomar secuencias de hasta 5994 y 12282 nucleótidos respectivamente si no hay "N" en su interior.
La colección de modelos presentada en este repositorio está disponible en los espacios Huggingface de Instadeep aquí: ¡El espacio del nucleotide transformer y el espacio nucleotide transformer agro!
Agradecemos a Maša Roller, así como a los miembros del Rostlab, en particular a Tobias Olenyi, Ivan Koludarov y Burkhard Rost, por las discusiones constructivas que ayudaron a identificar direcciones de investigación interesantes. Además, agradecemos a todos aquellos que depositan datos experimentales en bases de datos públicas, a quienes mantienen estas bases de datos y a quienes ponen a disposición de forma gratuita métodos analíticos y predictivos. También agradecemos al equipo de desarrollo de Jax.
Si encuentra útil este repositorio en su trabajo, agregue una cita relevante a cualquiera de nuestros artículos asociados:
El papel nucleotide transformer :
@article { dalla2023nucleotide ,
title = { The nucleotide transformer : Building and Evaluating Robust Foundation Models for Human Genomics } ,
author = { Dalla-Torre, Hugo and Gonzalez, Liam and Mendoza Revilla, Javier and Lopez Carranza, Nicolas and Henryk Grywaczewski, Adam and Oteri, Francesco and Dallago, Christian and Trop, Evan and Sirelkhatim, Hassan and Richard, Guillaume and others } ,
journal = { bioRxiv } ,
pages = { 2023--01 } ,
year = { 2023 } ,
publisher = { Cold Spring Harbor Laboratory }
}Papel nucleotide transformer agronucleótidos:
@article { mendoza2024foundational ,
title = { A foundational large language model for edible plant genomes } ,
author = { Mendoza-Revilla, Javier and Trop, Evan and Gonzalez, Liam and Roller, Ma{v{s}}a and Dalla-Torre, Hugo and de Almeida, Bernardo P and Richard, Guillaume and Caton, Jonathan and Lopez Carranza, Nicolas and Skwark, Marcin and others } ,
journal = { Communications Biology } ,
volume = { 7 } ,
number = { 1 } ,
pages = { 835 } ,
year = { 2024 } ,
publisher = { Nature Publishing Group UK London }
}Papel segmentoNT
@article { de2024segmentnt ,
title = { SegmentNT: annotating the genome at single-nucleotide resolution with DNA foundation models } ,
author = { de Almeida, Bernardo P and Dalla-Torre, Hugo and Richard, Guillaume and Blum, Christopher and Hexemer, Lorenz and Gelard, Maxence and Pandey, Priyanka and Laurent, Stefan and Laterre, Alexandre and Lang, Maren and others } ,
journal = { bioRxiv } ,
pages = { 2024--03 } ,
year = { 2024 } ,
publisher = { Cold Spring Harbor Laboratory }
}Si tiene alguna pregunta o comentario sobre el código y los modelos, no dude en comunicarse con nosotros.
¡Gracias por su interés en nuestro trabajo!


