BRAKER
v3.0.8
Aquí hay una grabación de la primera sesión del taller BGA23 en Braker. Si aprender videos es fácil para usted, considere ver eso: https://www.youtube.com/watch?v=uxtkj4mukyg
Braker3 ahora está en https://usegalaxy.eu/
Tsebra y Braker3 Relacionado:
Braker y Augustus relacionado:
Relacionado Genemark:
Mark Borodovsky, Georgia Tech, EE. UU., [email protected]
Tomas Bruna, Conjunto Genome Institute, EE. UU., [email protected]
Alexandre Lomsazde, Georgia Tech, EE. UU., [email protected]
[A] Universidad de Greifswald, Instituto de Matemáticas e Informática, Walther-Rathenau-Str. 47, 17489 Greifswald, Alemania
[B] Universidad de Greifswald, Centro de Genómica Funcional de Microbios, Felix-Hausdorff-Str. 8, 17489 Greifswald, Alemania
[C] Conjunto Georgia Tech y Emory University Wallace H Coulter Departamento de Ingeniería Biomédica, 30332 Atlanta, EE. UU.
[D] Escuela de Ciencia e Ingeniería Computacional, 30332 Atlanta, EE. UU.
[E] Instituto de Física y Tecnología de Moscú, Región de Moscú 141701, Dolgoprudny, Rusia
![Braker2-Team-2 [Fig10]](https://images.downcodes.com/uploads/20250214/img_67aee79a0cb7530.png)
![Braker2-Team-1 [Fig11]](https://images.downcodes.com/uploads/20250214/img_67aee79a0d1eb31.png)
![Braker2-Team-3 [Fig12]](https://images.downcodes.com/uploads/20250214/img_67aee79a0da9c32.png)
![Braker2-Team-4 [Fig13]](https://images.downcodes.com/uploads/20250214/img_67aee79a0e49933.png)
Figura 1: Autores actuales de los maestros, de izquierda a derecha: Mario Stanke, Alexandre Lomsadze, Katharina J. Hoff, Tomas Bruna, Lars Gabriel y Mark Borodovsky. Reconocemos que una comunidad más grande de científicos contribuyó al Código Braker (por ejemplo, a través de solicitudes de extracción).
El desarrollo de Braker1, Braker2 y Braker3 fue apoyado por los Institutos Nacionales de Salud (NIH) [GM128145 a MB y MS]. El desarrollo de Braker3 fue parcialmente financiado por la competencia de datos del proyecto otorgada a KJH y MS por el gobierno de Mecklenburg-VorPommern, Alemania.
El selector de transcripción para Braker (TSEBRA) está disponible en https://github.com/gaius-augustus/tsebra.
Genemark-ETP, uno de los buscadores de genes en el núcleo de Braker, está disponible en https://github.com/gatech-genemark/genemark-etp.
Augustus, el segundo buscador de genes en el núcleo de Braker, está disponible en https://github.com/gaius-augustus/augustus.
Galba, un spin-off de Braker Pipeline para usar Miniprot o Genomethreader para generar genes de entrenamiento, está disponible en https://github.com/gaius-augustus/galba.
El número rápido de genomas secuenciados requiere métodos completamente automatizados para la anotación precisa de la estructura génica. Con este objetivo en mente, hemos desarrollado Braker1 R1 R0 , una combinación de genemark-ET R2 y Augustus R3, R4 , que utiliza datos genómicos y de ARN-seq para generar automáticamente anotaciones de estructura genética completa en genoma novedoso.
Sin embargo, la calidad de los datos de ARN-seq que están disponibles para anotar un genoma novedoso es variable, y en algunos casos, los datos de RNA-seq no están disponibles en absoluto.
Braker2 es una extensión de Braker1 que permite el entrenamiento completamente automatizado de las herramientas de predicción de genes Genemark-ES/ET/EP/ETP R14, R15, R17, F1 y Augustus de la información de homología de RNA-seq y/o proteínas, y eso integra el Evidencia extrínseca de la información de homología de ARN-seq y proteína en la predicción .
A diferencia de otros métodos disponibles que dependen de la información de homología de proteínas, Braker2 alcanza una alta precisión de predicción genética incluso en ausencia de la anotación de especies muy relacionadas y en ausencia de datos de ARN-seq.
Braker3 es la última tubería en Braker Suite. Permite el uso de datos de ARN-seq y proteínas en una tubería totalmente automatizada para entrenar y predecir genes altamente confiables con Genemark-ETP y Augustus. El resultado de la tubería es el conjunto de genes combinado de ambas herramientas de predicción de genes, que solo contiene genes con un apoyo muy alto de evidencia extrínseca.
En esta guía del usuario, nos referiremos a Braker1, Braker2 y Braker3 simplemente como Braker porque son ejecutados por el mismo script ( braker.pl ).
Use un conjunto de genoma de alta calidad. Si tiene una gran cantidad de andamios muy cortos en su ensamblaje del genoma, esos andamios cortos probablemente aumentarán drásticamente el tiempo de ejecución, pero no aumentarán la precisión de la predicción.
Use nombres de andamio simples en el archivo del genoma (por ejemplo >contig1 funcionará mejor que >contig1my custom species namesome putative function /more/information/ and lots of special characters %&!*(){} ). Simplemente simples los nombres de andamio en todos sus archivos FASTA antes de ejecutar cualquier programa de alineación.
Para predecir genes con precisión en un genoma novedoso, el genoma debe enmascararse para repeticiones. Esto evitará la predicción de estructuras genéticas falsas positivas en regiones repetitivas y de baja complejidad. El enmascaramiento repetido también es esencial para mapear datos de RNA-seq a un genoma con algunas herramientas (otros mapeadores de ARN-seq, como HISAT2, ignoran la información de enmascaramiento). En el caso de Genemark-ES/ET/EP/ETP y Augustus, Softmasking (es decir, colocar regiones repetidas en letras minúsculas y todas las demás regiones en letras de mayúsculas) conduce a mejores resultados que el rígido (es decir, reemplazar letras en las regiones repetitivas por letra N para nucleótido desconocido).
Muchos genomas tienen estructuras genéticas que se predecirán con precisión con los parámetros estándar de Genemark-ES/ET/EP/ETP y Augustus dentro de Braker. Sin embargo, algunos genomas tienen características específicas de clado, es decir, un modelo especial de punto de ramificación en hongos o patrones no estándar del sitio de empalme. Lea la sección de opciones [opciones] para determinar si alguna de las opciones personalizadas puede mejorar la precisión de la predicción de genes en el genoma de su especie objetivo.
¡Vea siempre los resultados de la predicción del gen antes del uso adicional! Puede usar un navegador de genoma para la inspección visual de los modelos de genes en contexto con datos de evidencia extrínsecas. Braker admite la generación de centros de datos de seguimiento para el navegador Genome UCSC con Makehub para este propósito.
Braker presenta principalmente datos de evidencia extrínsecos semiupervisados (información de alineación empalmada de proteínas semi-supervisada (ARN-seq y/o información de alineación empalmada de proteínas) del entrenamiento respaldado de Genemark-ES/ET/EP/ETP [F1] y el entrenamiento posterior de Augustus con integración de evidencia extrínseca en la final paso de predicción de genes. Sin embargo, ahora hay una serie de tuberías adicionales incluidas en Braker. A continuación, damos una visión general de los posibles archivos de entrada y tuberías:
![Braker2-Main-A [Fig1]](https://images.downcodes.com/uploads/20250214/img_67aee79a0eaf534.png)
Figura 2: Beerelina A: Entrenamiento Genemark-ES en datos del genoma, solo; ab initio Predicción del gen con Augustus
![Braker2-Main-B [Fig2]](https://images.downcodes.com/uploads/20250214/img_67aee79a0f13f35.png)
Figura 3: Pipeline Beer B: Training Genemark-ET compatible con la información de alineación empalmada de ARN-seq, predicción con Augustus con la misma información de alineación empalmada.
![Braker2-Main-C [Fig3]](https://images.downcodes.com/uploads/20250214/img_67aee79a0fa1036.png)
Figura 4: Pipeline de cerebro C: Entrenamiento de la marca Genemark+ sobre alineación de proteínas empalmada, Información de inicio y detención, predicción con Augustus con esa misma información, además de sugerencias CDSSPART encadenadas. Las proteínas utilizadas aquí pueden ser de una distancia evolutiva al organismo objetivo.
![Braker3-Main-A [Fig4]](https://images.downcodes.com/uploads/20250214/img_67aee79a1010b37.png)
Figura 5: Pipeline de Braker D: si es necesario, descargue y alineación de conjuntos de ARN-seq para la especie objetivo. Entrenamiento de la marca genemática compatible con las alineaciones de ARN-seq y una gran base de datos de proteínas (las proteínas pueden tener una distancia evolutiva). Posteriormente, el entrenamiento y la predicción de Augustus utilizando la misma información extrínseca junto con los resultados de Genemark-ETP. La predicción final es la combinación Tsebra de los resultados de Augustus y Genemark-ETP.
Somos conscientes de que la instalación "manual" de Braker3 y todas sus dependencias es tediosa y realmente desafiante sin permisos de raíz. Por lo tanto, proporcionamos un contenedor Docker que se ha desarrollado para ejecutarse con singularidad. Toda la información en este contenedor se puede encontrar en https://hub.docker.com/r/teambraker/braker3
En resumen, construirlo de la siguiente manera:
singularity build braker3.sif docker://teambraker/braker3:latest
Ejecutar con:
singularity exec braker3.sif braker.pl
Prueba con:
singularity exec -B $PWD:$PWD braker3.sif cp /opt/BRAKER/example/singularity-tests/test1.sh .
singularity exec -B $PWD:$PWD braker3.sif cp /opt/BRAKER/example/singularity-tests/test2.sh .
singularity exec -B $PWD:$PWD braker3.sif cp /opt/BRAKER/example/singularity-tests/test3.sh .
export BRAKER_SIF=/your/path/to/braker3.sif # may need to modify
bash test1.sh
bash test2.sh
bash test3.sh
Pocos usuarios desean ejecutar su análisis dentro de Docker (ya que se requieren permisos de raíz). Sin embargo, si ese es su objetivo, puede ejecutar y probar el contenedor de la siguiente manera
sudo docker run --user 1000:100 --rm -it teambraker/braker3:latest bash
bash /opt/BRAKER/example/docker-tests/test1.sh # BRAKER1
bash /opt/BRAKER/example/docker-tests/test2.sh # BRAKER2
bash /opt/BRAKER/example/docker-tests/test3.sh # BRAKER3
Buena suerte ;-)
$PATH pueden conducir a interferencias imprevistas, causando fallas en el programa. Mueva todas las versiones genemarias más antiguas de su $PATH (también, por ejemplo, la marca Genemark en ProtHint/dependencies ).
En el momento del lanzamiento, esta versión de Braker se probó con:
Augustus 3.5.0 F2
Genemark-ETP (Fuente ver Dockerfile)
Bamtools 2.5.1 R5
SamTools 1.7-4-G93586ed R6
Spaln 2.3.3d R8, R9, R10
NCBI BLAST+ 2.2.31+ R12, R13
Diamante 0.9.24
CDBFASTA 0.99
Cdbyank 0.981
Gushr 1.0.0
SRA Toolkit 3.00 R14
HISAT2 2.2.1 R15
Bedtools 2.30 R16
StringTie2 2.2.1 R17
GFFREAD 0.12.7 R18
complacesm 0.2.5 R27
Ejecutar Braker requiere un sistema Linux con bash y Perl. Además, Braker requiere que se instalen los siguientes módulos CPAN-PERL:
File::Spec::Functions
Hash::Merge
List::Util
MCE::Mutex
Module::Load::Conditional
Parallel::ForkManager
POSIX
Scalar::Util::Numeric
YAML
Math::Utils
File::HomeDir
Para Genemark-ETP, se usa cuando se suministran proteínas y ARN-seq:
YAML::XSData::DumperThread::Queuethreads En Ubuntu, por ejemplo, instale los módulos con cpanminus f4 : sudo cpanm Module::Name , por ejemplo, sudo cpanm Hash::Merge .
Braker también usa un módulo Perl helpMod_braker.pm que no está disponible en CPAN. Este módulo es parte de la liberación de Braker y no requiere una instalación separada.
Si no tiene permisos raíz en la máquina Linux, intente configurar un entorno Anaconda (https://www.anaconda.com/distribution/) de la siguiente manera:
wget https://repo.anaconda.com/archive/Anaconda3-2018.12-Linux-x86_64.sh
bash bin/Anaconda3-2018.12-Linux-x86_64.sh # do not install VS (needs root privileges)
conda install -c anaconda perl
conda install -c anaconda biopython
conda install -c bioconda perl-app-cpanminus
conda install -c bioconda perl-file-spec
conda install -c bioconda perl-hash-merge
conda install -c bioconda perl-list-util
conda install -c bioconda perl-module-load-conditional
conda install -c bioconda perl-posix
conda install -c bioconda perl-file-homedir
conda install -c bioconda perl-parallel-forkmanager
conda install -c bioconda perl-scalar-util-numeric
conda install -c bioconda perl-yaml
conda install -c bioconda perl-class-data-inheritable
conda install -c bioconda perl-exception-class
conda install -c bioconda perl-test-pod
conda install -c bioconda perl-file-which # skip if you are not comparing to reference annotation
conda install -c bioconda perl-mce
conda install -c bioconda perl-threaded
conda install -c bioconda perl-list-util
conda install -c bioconda perl-math-utils
conda install -c bioconda cdbtools
conda install -c eumetsat perl-yaml-xs
conda install -c bioconda perl-data-dumper
Posteriormente, instale Braker y otro software "como de costumbre" mientras está en su entorno de condena. Nota: Hay un paquete BioConda Braker y un paquete BioConda Augustus. Ellos trabajan. Pero generalmente se quedan atrás del código de desarrollo de ambas herramientas en GitHub. Por lo tanto, recomendamos la instalación manual y el uso de las fuentes más últimas.
Braker es una colección de guiones de Perl y Python y un módulo de Perl. El script principal que se llamará para ejecutar Braker es braker.pl . Los componentes adicionales de Perl y Python son:
align2hints.pl
filterGenemark.pl
filterIntronsFindStrand.pl
startAlign.pl
helpMod_braker.pm
findGenesInIntrons.pl
downsample_traingenes.pl
ensure_n_training_genes.py
get_gc_content.py
get_etp_hints.py
Todos los scripts (archivos que terminan con *.pl y *.py ) que son parte de Braker deben ser ejecutables para ejecutar Braker. Este ya debería ser el caso si descarga Braker de GitHub. La ejecutabilidad puede sobrescribirse si por ejemplo, transferir al Braker en un palo USB a otra computadora. Para verificar si los archivos requeridos son ejecutables, ejecute el siguiente comando en el directorio que contiene scripts de Braker Perl:
ls -l *.pl *.py
La salida debe ser similar a esta:
-rwxr-xr-x 1 katharina katharina 18191 Mai 7 10:25 align2hints.pl
-rwxr-xr-x 1 katharina katharina 6090 Feb 19 09:35 braker_cleanup.pl
-rwxr-xr-x 1 katharina katharina 408782 Aug 17 18:24 braker.pl
-rwxr-xr-x 1 katharina katharina 5024 Mai 7 10:25 downsample_traingenes.pl
-rwxr-xr-x 1 katharina katharina 5024 Mai 7 10:23 ensure_n_training_genes.py
-rwxr-xr-x 1 katharina katharina 4542 Apr 3 2019 filter_augustus_gff.pl
-rwxr-xr-x 1 katharina katharina 30453 Mai 7 10:25 filterGenemark.pl
-rwxr-xr-x 1 katharina katharina 5754 Mai 7 10:25 filterIntronsFindStrand.pl
-rwxr-xr-x 1 katharina katharina 7765 Mai 7 10:25 findGenesInIntrons.pl
-rwxr-xr-x 1 katharina katharina 1664 Feb 12 2019 gatech_pmp2hints.pl
-rwxr-xr-x 1 katharina katharina 2250 Jan 9 13:55 log_reg_prothints.pl
-rwxr-xr-x 1 katharina katharina 4679 Jan 9 13:55 merge_transcript_sets.pl
-rwxr-xr-x 1 katharina katharina 41674 Mai 7 10:25 startAlign.pl
Es importante que el x en -rwxr-xr-x esté presente para cada script. Si ese no es el caso, ejecute
`chmod a+x *.pl *.py`
Para cambiar los atributos de archivo.
Puede que sea útil agregar el directorio en el que los scripts de Braker Perl residen a su variable de entorno $PATH . Para una sola sesión de Bash, ingrese:
PATH=/your_path_to_braker/:$PATH
export PATH
Para que esta modificación $PATH esté disponible para todas las sesiones de bash, agregue las líneas anteriores a un script de inicio (por ejemplo, ~/.bashrc ).
Braker recurre a varias herramientas de software de bioinformática que no forman parte de Braker. Algunas herramientas son obligatorias, es decir, Braker no se ejecutará en absoluto si estas herramientas no están presentes en su sistema. Otras herramientas son opcionales. Instale todas las herramientas necesarias para ejecutar Braker en el modo de su elección.
Descargue genemark-etp f1 de http://github.com/gatech-genemark/genemark-etp o https://topaz.gatech.edu/genemark/etp.for_braker.tar.gz. Desempacar e instalar Genemark-ETP como se describe en el archivo README de Genemark-ETP.
Si ya está contenido en su variable $PATH , Braker adivinará la ubicación de gmes_petap.pl o gmetp.pl automáticamente. De lo contrario, Braker puede encontrar ejecutables Genemark-ES/ET/EP/ETP, ya sea ubicándolos en una variable de entorno, GENEMARK_PATH , o tomando un argumento de línea de comandos ( --GENEMARK_PATH=/your_path_to_GeneMark_executables/ ).
Para establecer la variable de entorno para su sesión de Bash actual, escriba:
export GENEMARK_PATH=/your_path_to_GeneMark_executables/
Agregue las líneas anteriores a un script de inicio (p. ~/.bashrc ) para que esté disponible para todas las sesiones de bash.
Los scripts de Perl dentro de Genemark-ES/ET/EP/ETP están configurados con la ubicación PERL predeterminada AT /usr/bin/perl .
Si está ejecutando Genemark-ES/ET/EP/ETP en un entorno de Anaconda (o desea usar Perl de la variable $PATH por cualquier otra razón), modifique el shebang de todos los scripts genemark-es/ET/EP/ETP con El siguiente comando ubicado dentro de la carpeta Genemark-ES/ET/EP/ETP:
perl change_path_in_perl_scripts.pl "/usr/bin/env perl"
Puede verificar si Genemark-ES/ET/EP se instala correctamente ejecutando el directorio check_install.bash y/o ejecutando ejemplos en el directorio GeneMark-E-tests .
Genemark-ETP es descendente compatible, es decir, cubre la funcionalidad de Genemark-Ep y Genemark-ET en Braker, también.
Descargue Augustus desde su rama maestra en https://github.com/gaius-augustus/augustus. Desempaque a Augustus e instale Augustus según Augustus README.TXT . ¡No use versiones anticuadas de Augustus de otras fuentes, por ejemplo, el paquete Debian o el paquete Bioconda! El que Beer depende en particular en un directorio de Augustus/Scripts actualizado, y otras fuentes a menudo se quedan atrás.
Debe compilar Augustus en su propio sistema para evitar problemas con las versiones de las bibliotecas utilizadas por Augustus. Las instrucciones de compilación se proporcionan en el archivo Augustus README.TXT ( Augustus/README.txt ).
Augustus está formado por augustus , la herramienta de predicción de genes, herramientas adicionales de C ++ ubicadas en Augustus/auxprogs y Scripts Perl ubicados en Augustus/scripts . Los scripts de Perl deben ser ejecutables (consulte las instrucciones en la sección Componentes de Braker.
La herramienta C ++ bam2hints es un componente esencial de Braker cuando se ejecuta con RNA-seq. Las fuentes se encuentran en Augustus/auxprogs/bam2hints . Asegúrese de compilar bam2hints en su sistema (debe compilarse automáticamente cuando Augustus se compile, pero en caso de problemas con bam2hints , lea las instrucciones de solución de problemas en Augustus/auxprogs/bam2hints/README ).
Dado que Braker es una tubería que entrena a Augustus, IE escribe archivos de parámetros específicos de especies, Braker necesita escribir acceso al directorio de configuración de Augustus que contiene dichos archivos ( Augustus/config/ ). Si instala Augustus a nivel mundial en su sistema, la carpeta config generalmente no será redactada por todos los usuarios. Haga que el directorio donde config reside recursivamente es escrito para los usuarios de Augustus, o copie la carpeta config/ (recursivamente) a una ubicación donde los usuarios tienen permiso de escritura.
Augustus localizará la carpeta de config buscando una variable de entorno $AUGUSTUS_CONFIG_PATH . Si no se establece la variable de entorno $AUGUSTUS_CONFIG_PATH , entonces Braker buscará en la ruta ../config en relación con el directorio en el que encuentra un ejecutable de Augustus. Alternativamente, puede suministrar la variable como un argumento de línea de comando a Braker ( --AUGUSTUS_CONFIG_PATH=/your_path_to_AUGUSTUS/Augustus/config/ ). Recomendamos que exporte la variable, por ejemplo, para su sesión de Bash actual:
export AUGUSTUS_CONFIG_PATH=/your_path_to_AUGUSTUS/Augustus/config/
Para que la variable esté disponible para todas las sesiones de bash, agregue la línea anterior a un script de inicio, por ejemplo, ~/.bashrc .
Eche un vistazo al DockerFile en caso de que desee instalar Augustus como paquete Debian. Una serie de scripts deben ser parcheados, entonces.
¡Braker espera todo el directorio config de Augustus en $AUGUSTUS_CONFIG_PATH , es decir, las species de subcarpetas con su contenido (al menos generic ) y extrinsic ! Proporcionar una carpeta de escritura pero vacía a $AUGUSTUS_CONFIG_PATH no funcionará para Braker. Si necesita separar Augustus Binary y $AUGUSTUS_CONFIG_PATH , le recomendamos que copie recursivamente el contenido de configuración no escrita en una ubicación de escritura.
Si tiene una instalación en todo el sistema de Augustus AT /usr/bin/augustus , una copia no escrita de config se encuentra AT /usr/bin/augustus_config/ . La carpeta /home/yours/ es una escritura para ti. Copie con el siguiente comando (y establezca adicionalmente las variables requeridas):
cp -r /usr/bin/Augustus/config/ /home/yours/
export AUGUSTUS_CONFIG_PATH=/home/yours/augustus_config
export AUGUSTUS_BIN_PATH=/usr/bin
export AUGUSTUS_SCRIPTS_PATH=/usr/bin/augustus_scripts
Agregar directorios de los binarios y scripts de Augustus a su variable $PATH permite que su sistema localice estas herramientas, automáticamente. No es un requisito para ejecutar Braker para hacer esto, porque Braker intentará adivinarlos desde la ubicación de otra variable de entorno ( $AUGUSTUS_CONFIG_PATH ), o ambos directorios pueden suministrarse como argumentos de línea de comandos a braker.pl , pero recomendamos a Agrégalos a su variable $PATH . Para su sesión de Bash actual, escriba:
PATH=:/your_path_to_augustus/bin/:/your_path_to_augustus/scripts/:$PATH
export PATH
Para todas sus sesiones de Bash, agregue las líneas anteriores a un script de inicio (por ejemplo, ~/.bashrc ).
En Ubuntu, Python3 generalmente se instala de forma predeterminada, python3 estará en su variable $PATH , por defecto, y Braker la localizará automáticamente. Sin embargo, tiene la opción de especificar la ubicación binaria python3 de otras dos maneras:
Exportar una variable de entorno $PYTHON3_PATH , por ejemplo, en su archivo ~/.bashrc :
export PYTHON3_PATH=/path/to/python3/
Especifique la opción Línea de comando --PYTHON3_PATH=/path/to/python3/ To braker.pl .
Descargue BamTools (por ejemplo git clone https://github.com/pezmaster31/bamtools.git ). Instale bamtools escribiendo lo siguiente en su carcasa:
cd your-bamtools-directory mkdir build cd build cmake .. make
Si ya está en su variable $PATH , Braker encontrará BamTools, automáticamente. De lo contrario, Braker puede ubicar el binario BamTools utilizando una variable de entorno $BAMTOOLS_PATH , o tomando un argumento de línea de comando ( --BAMTOOLS_PATH=/your_path_to_bamtools/bin/ F6 ). Para establecer la variable de entorno, por ejemplo, para su sesión de bash actual, escriba:
export BAMTOOLS_PATH=/your_path_to_bamtools/bin/
Agregue la línea anterior a un script de inicio (p. ~/.bashrc ) para establecer la variable de entorno para todas las sesiones BASH.
Puede usar NCBI Blast+ o Diamond para eliminar los genes de entrenamiento redundantes. No necesitas ambas herramientas. Si el diamante está presente, se preferirá porque es mucho más rápido.
Obtenga y desempaquete el diamante de la siguiente manera:
wget http://github.com/bbuchfink/diamond/releases/download/v0.9.24/diamond-linux64.tar.gz
tar xzf diamond-linux64.tar.gz
Si ya está en su variable $PATH , Braker encontrará Diamond, automáticamente. De lo contrario, Braker puede localizar el binario de diamantes utilizando una variable de entorno $DIAMOND_PATH , o tomando un argumento de línea de comando ( --DIAMOND_PATH=/your_path_to_diamond ). Para establecer la variable de entorno, por ejemplo, para su sesión de bash actual, escriba:
export DIAMOND_PATH=/your_path_to_diamond/
Agregue la línea anterior a un script de inicio (p. ~/.bashrc ) para establecer la variable de entorno para todas las sesiones BASH.
Si decide BLAST+, instale NCBI BLAST+ con sudo apt-get install ncbi-blast+ .
Si ya está en su variable $PATH , Braker encontrará BLASTP, automáticamente. De lo contrario, Braker puede localizar el binario BLASTP utilizando una variable de entorno $BLAST_PATH , o tomando un argumento de línea de comando ( --BLAST_PATH=/your_path_to_blast/ ). Para establecer la variable de entorno, por ejemplo, para su sesión de bash actual, escriba:
export BLAST_PATH=/your_path_to_blast/
Agregue la línea anterior a un script de inicio (p. ~/.bashrc ) para establecer la variable de entorno para todas las sesiones BASH.
Genemark-ETP requiere las siguientes herramientas e intentará ubicarlas en su variable $PATH . Así que asegúrese de agregar su ubicación a su $PATH , por ejemplo:
export PATH=$PATH:/your/path/to/Tool
Para todas las herramientas a continuación, agregue la línea anterior a un script de inicio (por ejemplo, ~/.bashrc ) para extender su variable $PATH para todas las sesiones de bash.
¡Estas herramientas de software solo son obligatorias si ejecuta Braker con RNA-seq y datos de proteínas!
StringTie2 es utilizado por Genemark-ETP para ensamblar alineaciones de ARN-seq alineadas. Se puede descargar una versión precompilada de StringTie2 de https://ccb.jhu.edu/software/stringtie/#install.
El paquete de software Bedtools es requerido por Genemark-ETP si desea ejecutar Braker con datos de ARN-seq y proteína. Puede descargar BedTools de https://github.com/arq5x/bedtools2/releases. Aquí, puede descargar una versión precompilada bedtools.static.binary , por ejemplo
wget https://github.com/arq5x/bedtools2/releases/download/v2.30.0/bedtools.static.binary
mv bedtools.static.binary bedtools
chmod a+x
O puede descargar bedtools-2.30.0.tar.gz y compilarlo desde la fuente usando make , EG
wget https://github.com/arq5x/bedtools2/releases/download/v2.30.0/bedtools-2.30.0.tar.gz
tar -zxvf bedtools-2.30.0.tar.gz
cd bedtools2
make
Consulte https://bedtools.readthedocs.io/en/latest/content/installation.html para obtener más información.
GFFREAD es un software de utilidad requerido por Genemark-ETP. Se puede descargar desde https://github.com/gpertea/gffread/releases/download/v0.12.7/gffread-0.12.7.linux_x86_64.tar.gz e instalado con make ,
wget https://github.com/gpertea/gffread/releases/download/v0.12.7/gffread-0.12.7.Linux_x86_64.tar.gz
tar xzf gffread-0.12.7.Linux_x86_64.tar.gz
cd gffread-0.12.7.Linux_x86_64
make
SamTools no es necesario para ejecutar Braker sin Genemark-ETP si todos sus archivos están formateados, correctamente (es decir, todas las secuencias deberían tener nombres FASTA cortos y únicos). Si no está seguro de si todos sus archivos están confundidos correctamente, podría ser útil tener a SamTools instalado porque Braker puede solucionar automáticamente ciertos problemas de formato mediante el uso de SamTools.
Como un requisito previo para SamTools, descargue e instale htslib (por ejemplo, git clone https://github.com/samtools/htslib.git , siga la documentación htslib para la instalación).
Descargue e instale SamTools (por ejemplo, git clone git://github.com/samtools/samtools.git ), posteriormente siga la documentación de SamTools para su instalación).
Si ya está en su variable $PATH , Braker encontrará SamTools, automáticamente. De lo contrario, Braker puede encontrar SamTools, ya sea tomando un argumento de línea de comando ( --SAMTOOLS_PATH=/your_path_to_samtools/ ), o utilizando una variable de entorno $SAMTOOLS_PATH . Para exportar la variable, por ejemplo, para su sesión de Bash actual, escriba:
export SAMTOOLS_PATH=/your_path_to_samtools/
Agregue la línea anterior a un script de inicio (p. ~/.bashrc ) para establecer la variable de entorno para todas las sesiones BASH.
Si se instala Biopython, Braker puede generar archivos FASTA con secuencias de codificación y secuencias de proteínas predichas por Augustus y generar centros de datos de seguimiento para la visualización de un Braker ejecutado con Makehub R16 . Estos son pasos opcionales. El primero se puede deshabilitar con el indicador de la línea de comandos --skipGetAnnoFromFasta , el segundo puede activarse utilizando las opciones de línea de comandos --makehub [email protected] , Biopython no es necesario si ninguno de estos pasos opcionales se realizará.
En Ubuntu, instale el Administrador de paquetes Python3 con:
`sudo apt-get install python3-pip`
Luego, instale Biopython con:
`sudo pip3 install biopython`
CDBFASTA y CDBYANK son requeridos por Braker para corregir los genes de Augustus con codones de parada de cuadro (codones de parada empalmados) utilizando el script Augustus Fix_in_frame_stop_codon_genes.py. Esto se puede omitir con --skip_fixing_broken_genes .
En Ubuntu, instale CDBFastA con:
sudo apt-get install cdbfasta
Para otros sistemas,, por ejemplo, puede obtener CDBFastA de https://github.com/gpertea/cdbfasta, por ejemplo:
git clone https://github.com/gpertea/cdbfasta.git
cd cdbfasta
make all
En Ubuntu, CDBFastA y CDByank estarán en su variable $PATH después de la instalación, y Braker los localizará automáticamente. Sin embargo, tiene la opción de especificar la ubicación binaria cdbfasta y cdbyank de otras dos maneras:
$CDBTOOLS_PATH , por ejemplo, en su archivo ~/.bashrc : export CDBTOOLS_PATH=/path/to/cdbtools/
--CDBTOOLS_PATH=/path/to/cdbtools/ TO braker.pl . Nota: El soporte de Spaln independiente (OUSide of Prothint) dentro de Braker está en desuso.
Esta herramienta es necesaria si ejecuta protonos o si desea ejecutar alineaciones de proteínas a genoma con Braker usando Spaln fuera del Prótes. El uso de spaln fuera del prothint es un enfoque adecuado solo si una especie anotada de corta distancia evolutiva a su genoma objetivo está disponible. Recomendamos ejecutar Spaln a través de Prothint para Braker. Prothint trae un binario spaln. Si eso no funciona en su sistema, descargue Spaln de https://github.com/ogotoh/spaln. Desempacar e instalar según spaln/doc/SpalnReadMe22.pdf .
Braker intentará localizar el ejecutable de SPALN utilizando una variable de entorno $ALIGNMENT_TOOL_PATH . Alternativamente, esto se puede suministrar como argumento de línea de comandos ( --ALIGNMENT_TOOL_PATH=/your/path/to/spaln ).
Esta herramienta solo se requiere si desea agregar UTRS (de los datos de RNA-seq) a los genes predichos o si desea entrenar los parámetros de UTR para Augustus y predecir los genes con UTRS. En cualquier caso, GushR requiere la entrada de datos de ARN-seq.
Gushr está disponible para descargar en https://github.com/gaius-augustus/gushr. Obtenerlo escribiendo:
git clone https://github.com/Gaius-Augustus/GUSHR.git
Gushr ejecuta un archivo Gemoma JAR R19, R20, R21 , y este archivo JAR requiere Java 1.8. En Ubuntu, puede instalar Java 1.8 con el siguiente comando:
sudo apt-get install openjdk-8-jdk
Si tiene varias versiones Java instaladas en su sistema, asegúrese de habilitar 1.8 antes de ejecutar Braker con Java ejecutando
sudo update-alternatives --config java
y seleccionar la versión correcta.
Si cambia --UTR=on , bamtowig.py requerirá las siguientes herramientas que se pueden descargar desde http://hgdownload.soe.ucsc.edu/admin/exe:
twobitinfo
fatotwobit
Es opcional instalar estas herramientas en su ruta $. Si no lo hace, y cambia --UTR=on , bamtowig.py los descargará automáticamente en el directorio de trabajo.
Si desea automaticaly generar un centro de datos de seguimiento de su ejecución de Braker, se requiere el software Makehub, disponible en https://github.com/gaius-augustus/makehub. Descargue el software (ya sea ejecutando git clone https://github.com/Gaius-Augustus/MakeHub.git , o eligiendo un lanzamiento de https://github.com/gaius-augustus/makehub/releases. Extraiga el lanzamiento. Paquete si descargó una versión (por ejemplo, unzip MakeHub.zip o tar -zxvf MakeHub.tar.gz .
Braker intentará localizar el script make_hub.py utilizando una variable de entorno $MAKEHUB_PATH . Alternativamente, esto se puede suministrar como argumento de línea de comandos ( --MAKEHUB_PATH=/your/path/to/MakeHub/ ). Braker también puede tratar de adivinar la ubicación de Makehub en su sistema.
Si desea que Braker descargue las bibliotecas RNA-Seq de la SRA de NCBI, se requiere el kit de herramientas SRA. Puede obtener una versión precompilada del kit de herramientas SRA de http://daehwankimlab.github.io/hisat2/download/#version-hisat2-221.
Braker intentará encontrar binarios ejecutables del SRA Toolkit (FastQ-Dump, Prepletch) utilizando una variable de entorno $SRATOOLS_PATH . Alternativamente, esto se puede suministrar como argumento de línea de comandos ( --SRATOOLS_PATH=/your/path/to/SRAToolkit/ ). Braker también puede intentar adivinar la ubicación del kit de herramientas SRA en su sistema si los ejecutables están en su variable $PATH .
Si desea usar lecturas de ARN-seq no alineadas, se requiere el software HISAT2 para asignarlos al genoma. Se puede descargar una versión precompilada de HISAT2 de http://daehwankimlab.github.io/hisat2/download/#version-hisat2-221.
Braker intentará encontrar binarios ejecutables de Hisat2 (Hisat2, Hisat2-Build) utilizando una variable de entorno $HISAT2_PATH . Alternativamente, esto se puede suministrar como argumento de línea de comandos ( --HISAT2_PATH=/your/path/to/HISAT2/ ). Braker también puede intentar adivinar la ubicación de HISAT2 en su sistema si los ejecutables están en su variable $PATH .
Si desea ejecutar Tsebra dentro de Braker en un modo de maximización de integridad de Busco, debe instalar Complesm.
wget https://github.com/huangnengCSU/compleasm/releases/download/v0.2.4/compleasm-0.2.4_x64-linux.tar.bz2
tar -xvjf compleasm-0.2.4_x64-linux.tar.bz2 &&
Agregue la carpeta resultante que se complete a su variable $PATH , por ejemplo:
export PATH=$PATH:/your/path/to/compleasm_kit
Complesm requiere pandas, que se pueden instalar con:
pip install pandas
Braker (Braker.PL) usa GetConf para ver cuántos hilos se pueden ejecutar en su sistema. En Ubuntu, puede instalarlo con:
sudo apt-get install libc-bin
A continuación, describimos las llamadas "típicas" de los exitivos para diferentes tipos de datos de entrada. En general, le recomendamos que ejecute a Braker en secuencias genómicas que han sido blandas para repeticiones. ¡Braker solo debe aplicarse a los genomas que han sido blandas para repeticiones!
This approach is suitable for genomes of species for which RNA-Seq libraries with good transcriptome coverage are available and for which protein data is not at hand. The pipeline is illustrated in Figure 2.
BRAKER has several ways to receive RNA-Seq data as input:
You can provide ID(s) of RNA-Seq libraries from SRA (in case of multiple IDs, separate them by comma) as argument to --rnaseq_sets_ids . The libraries belonging to the IDs are then downloaded automatically by BRAKER, eg:
braker.pl --species=yourSpecies --genome=genome.fasta
--rnaseq_sets_ids=SRA_ID1,SRA_ID2
You can use local FASTQ file(s) of unaligned reads as input. In this case, you have to provide BRAKER with the ID(s) of the RNA-Seq set(s) as argument to --rnaseq_sets_ids and the path(s) to the directories, where the FASTQ files are located as argument to --rnaseq_sets_dirs . For each ID ID , BRAKER will search in these directories for one FASTQ file named ID.fastq if the reads are unpaired, or for two FASTQ files named ID_1.fastq and ID_2.fastq if they are paired.
For example, if you have a paired library called 'SRA_ID1' and an unpaired library named 'SRA_ID2', you have to have a directory /path/to/local/fastq/files/ , where the files SRA_ID1_1.fastq , SRA_ID1_2.fastq , and SRA_ID2.fastq reside. Then, you could run BRAKER with following command:
braker.pl --species=yourSpecies --genome=genome.fasta
--rnaseq_sets_ids=SRA_ID1,SRA_ID2
--rnaseq_sets_dirs=/path/to/local/fastq/files/
There are two ways of supplying BRAKER with RNA-Seq data as bam file(s). First, you can do it in the same way as you would supply FASTQ file(s): Provide the ID(s)/name(s) of your bam file(s) as argument to --rnaseq_sets_ids and specify directories where the bam files reside with --rnaseq_sets_dirs . BRAKER will automatically detect that these ID(s) are bam and not FASTQ file(s), eg:
braker.pl --species=yourSpecies --genome=genome.fasta
--rnaseq_sets_ids=BAM_ID1,BAM_ID2
--rnaseq_sets_dirs=/path/to/local/bam/files/
Second, you can specify the paths to your bam file(s) directly, eg can either extract RNA-Seq spliced alignment information from bam files, or it can use such extracted information, directly.
braker.pl --species=yourSpecies --genome=genome.fasta
--bam=file1.bam,file2.bam
Please note that we generally assume that bam files were generated with HiSat2 because that is the aligner that would also be executed by BRAKER3 with fastq input. If you want for some reason to generate the bam files with STAR, use the option --outSAMstrandField intronMotif of STAR to produce files that are compatible wiht StringTie in BRAKER3.
In order to run BRAKER with RNA-Seq spliced alignment information that has already been extracted, run:
braker.pl --species=yourSpecies --genome=genome.fasta
--hints=hints1.gff,hints2.gff
The format of such a hints file must be as follows (tabulator separated file):
chrName b2h intron 6591 8003 1 + . pri=4;src=E
chrName b2h intron 6136 9084 11 + . mult=11;pri=4;src=E
...
The source b2h in the second column and the source tag src=E in the last column are essential for BRAKER to determine whether a hint has been generated from RNA-Seq data.
It is also possible to provide RNA-Seq sets in different ways for the same BRAKER run, any combination of above options is possible. It is not recommended to provide RNA-Seq data with --hints if you run BRAKER in ETPmode (RNA-Seq and protein data), because GeneMark-ETP won't use these hints!
This approach is suitable for genomes of species for which no RNA-Seq libraries are available. A large database of proteins (with possibly longer evolutionary distance to the target species) should be used in this case. This mode is illustrated in figure 9.
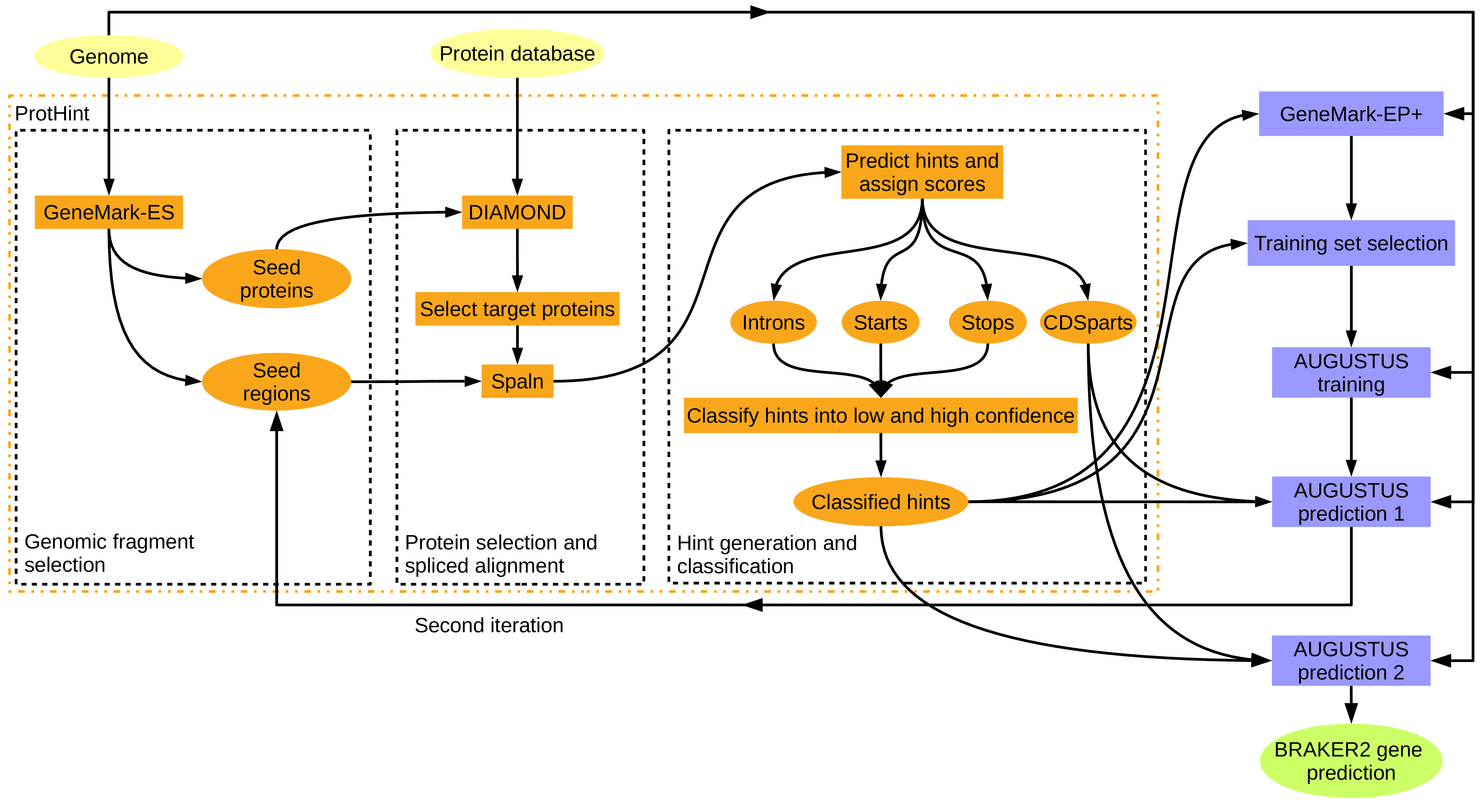
Figure 9: BRAKER with proteins of any evolutionary distance. ProtHint protein mapping pipelines is used to generate protein hints. ProtHint automatically determines which alignments are from close relatives, and which are from rather distant relatives.
For running BRAKER in this mode, type:
braker.pl --genome=genome.fa --prot_seq=proteins.fa
We recommend using OrthoDB as basis for proteins.fa . The instructions on how to prepare the input OrthoDB proteins are documented here: https://github.com/gatech-genemark/ProtHint#protein-database-preparation.
You can of course add additional protein sequences to that file, or try with a completely different database. Any database will need several representatives for each protein, though.
Instead of having BRAKER run ProtHint, you can also start BRAKER with hints already produced by ProtHint, by providing ProtHint's prothint_augustus.gff output:
braker.pl --genome=genome.fa --hints=prothint_augustus.gff
The format of prothint_augustus.gff in this mode looks like this:
2R ProtHint intron 11506230 11506648 4 + . src=M;mult=4;pri=4
2R ProtHint intron 9563406 9563473 1 + . grp=69004_0:001de1_702_g;src=C;pri=4;
2R ProtHint intron 8446312 8446371 1 + . grp=43151_0:001cae_473_g;src=C;pri=4;
2R ProtHint intron 8011796 8011865 2 - . src=P;mult=1;pri=4;al_score=0.12;
2R ProtHint start 234524 234526 1 + . src=P;mult=1;pri=4;al_score=0.08;
The prediction of all hints with src=M will be enforced. Hints with src=C are 'chained evidence', ie they will only be incorporated if all members of the group (grp=...) can be incorporated in a single transcript. All other hints have src=P in the last column. Supported features in column 3 are intron , start , stop and CDSpart .
If RNA-Seq (and only RNA-Seq) data is provided to BRAKER as a bam-file, and if the genome is softmasked for repeats, BRAKER can automatically train UTR parameters for AUGUSTUS. After successful training of UTR parameters, BRAKER will automatically predict genes including coverage information form RNA-Seq data. Example call:
braker.pl --species=yourSpecies --genome=genome.fasta
--bam=file.bam --UTR=on
Advertencias:
This feature is experimental!
--UTR=on is currently not compatible with bamToWig.py as released in AUGUSTUS 3.3.3; it requires the current development code version from the github repository (git clone https://github.com/Gaius-Augustus/Augustus.git).
--UTR=on increases memory consumption of AUGUSTUS. Carefully monitor jobs if your machine was close to maxing RAM without --UTR=on! Reducing the number of cores will also reduce RAM consumption.
UTR prediction sometimes improves coding sequence prediction accuracy, but not always. If you try this feature, carefully compare results with and without UTR parameters, afterwards (eg in UCSC Genome Browser).
For running BRAKER without UTR parameters, it is not very important whether RNA-Seq data was generated by a stranded protocol (because spliced alignments are 'artificially stranded' by checking the splice site pattern). However, for UTR training and prediction, stranded libraries may provide information that is valuable for BRAKER.
After alignment of the stranded RNA-Seq libraries, separate the resulting bam file entries into two files: one for plus strand mappings, one for minus strand mappings. Call BRAKER as follows:
braker.pl --species=yourSpecies --genome=genome.fasta
--bam=plus.bam,minus.bam --stranded=+,-
--UTR=on
You may additionally include bam files from unstranded libraries. Those files will not used for generating UTR training examples, but they will be included in the final gene prediction step as unstranded coverage information, example call:
braker.pl --species=yourSpecies --genome=genome.fasta
--bam=plus.bam,minus.bam,unstranded.bam
--stranded=+,-,. --UTR=on
Warning: This feature is experimental and currently has low priority on our maintenance list!
The native mode for running BRAKER with RNA-Seq and protein data. This will call GeneMark-ETP, which will use RNA-Seq and protein hints for training GeneMark-ETP. Subsequently, AUGUSTUS is trained on 'high-confindent' genes (genes with very high extrinsic evidence support) from the GeneMark-ETP prediction and a set of genes is predicted by AUGUSTUS. In a last step, the predictions of AUGUSTUS and GeneMark-ETP are combined using TSEBRA.
Alignment of RNA-Seq reads
GeneMark-ETP utilizes Stringtie2 to assemble RNA-Seq data, which requires that the aligned reads (BAM files) contain the XS (strand) tag for spliced reads. Therefore, if you align your reads with HISAT2, you must enable the --dta option, or if you use STAR, you must use the --outSAMstrandField intronMotif option. TopHat alignments include this tag by default.
To call the pipeline in this mode, you have to provide it with a protein database using --prot_seq (as described in BRAKER with protein data), and RNA-Seq data either by their SRA ID so that they are downloaded by BRAKER, as unaligned reads in FASTQ format, and/or as aligned reads in bam format (as described in BRAKER with RNA-Seq data). You could also specify already processed extrinsic evidence using the --hints option. However, this is not recommend for a normal BRAKER run in ETPmode, as these hints won't be used in the GeneMark-ETP step. Only use --hints when you want to skip the GenMark-ETP step!
Examples of how you could run BRAKER in ETPmode:
braker.pl --genome=genome.fa --prot_seq=orthodb.fa
--rnaseq_sets_ids=SRA_ID1,SRA_ID2
--rnaseq_sets_dirs=/path/to/local/RNA-Seq/files/
braker.pl --genome=genome.fa --prot_seq=orthodb.fa
--rnaseq_sets_ids=SRA_ID1,SRA_ID2,SRA_ID3
braker.pl --genome=genome.fa --prot_seq=orthodb.fa
--bam=/path/to/SRA_ID1.bam,/path/to/SRA_ID2.bam
A preliminary protocol for integration of assembled subreads from PacBio ccs sequencing in combination with short read Illumina RNA-Seq and protein database is described at https://github.com/Gaius-Augustus/BRAKER/blob/master/docs/long_reads/long_read_protocol .Maryland
We forked GeneMark-ETP and hard coded that StringTie will perform long read assembly in that particular version. If you want to use this 'fast-hack' version for BRAKER, you have to prepare the BAM file with long read to genome spliced alignments outside of BRAKER, eg:
T=48 # adapt to your number of threads
minimap2 -t${T} -ax splice:hq -uf genome.fa isoseq.fa > isoseq.sam
samtools view -bS --threads ${T} isoseq.sam -o isoseq.bam
Pull the adapted container:
singularity build braker3_lr.sif docker://teambraker/braker3:isoseq
Calling BRAKER3 with a BAM file of spliced-aligned IsoSeq Reads:
singularity exec -B ${PWD}:${PWD} braker3_lr.sif braker.pl --genome=genome.fa --prot_seq=protein_db.fa –-bam=isoseq.bam --threads=${T}
Warning Do NOT mix short read and long read data in this BRAKER/GeneMark-ETP variant!
Warning The accuracy of gene prediction here heavily depends on the depth of your isoseq data. We verified with PacBio HiFi reads from 2022 that given sufficient completeness of the assembled transcriptome you will reach similar results as with short reads. However, we also observed a drop in accuracy compared to short reads when using other long read data sets with higher error rates and less sequencing depth.
Please run braker.pl --help to obtain a full list of options.
Compute AUGUSTUS ab initio predictions in addition to AUGUSTUS predictions with hints (additional output files: augustus.ab_initio.* . This may be useful for estimating the quality of training gene parameters when inspecting predictions in a Browser.
One or several command line arguments to be passed to AUGUSTUS, if several arguments are given, separate them by whitespace, ie "--first_arg=sth --second_arg=sth" . This may be be useful if you know that gene prediction in your particular species benefits from a particular AUGUSTUS argument during the prediction step.
Specifies the maximum number of threads that can be used during computation. BRAKER has to run some steps on a single thread, others can take advantage of multiple threads. If you use more than 8 threads, this will not speed up all parallelized steps, in particular, the time consuming optimize_augustus.pl will not use more than 8 threads. However, if you don't mind some threads being idle, using more than 8 threads will speed up other steps.
GeneMark-ETP option: run algorithm with branch point model. Use this option if you genome is a fungus.
Use the present config and parameter files if they exist for 'species'; will overwrite original parameters if BRAKER performs an AUGUSTUS training.
Execute CRF training for AUGUSTUS; resulting parameters are only kept for final predictions if they show higher accuracy than HMM parameters. This increases runtime!
Change the parameter
Generate UTR training examples for AUGUSTUS from RNA-Seq coverage information, train AUGUSTUS UTR parameters and predict genes with AUGUSTUS and UTRs, including coverage information for RNA-Seq as evidence. This is an experimental feature!
If you performed a BRAKER run without --UTR=on, you can add UTR parameter training and gene prediction with UTR parameters (and only RNA-Seq hints) with the following command:
braker.pl --genome=../genome.fa --addUTR=on
--bam=../RNAseq.bam --workingdir=$wd
--AUGUSTUS_hints_preds=augustus.hints.gtf
--threads=8 --skipAllTraining --species=somespecies
Modify augustus.hints.gtf to point to the AUGUSTUS predictions with hints from previous BRAKER run; modify flaning_DNA value to the flanking region from the log file of your previous BRAKER run; modify some_new_working_directory to the location where BRAKER should store results of the additional BRAKER run; modify somespecies to the species name used in your previous BRAKER run.
Add UTRs from RNA-Seq converage information to AUGUSTUS gene predictions using GUSHR. No training of UTR parameters and no gene prediction with UTR parameters is performed.
If you performed a BRAKER run without --addUTR=on, you can add UTRs results of a previous BRAKER run with the following command:
braker.pl --genome=../genome.fa --addUTR=on
--bam=../RNAseq.bam --workingdir=$wd
--AUGUSTUS_hints_preds=augustus.hints.gtf --threads=8
--skipAllTraining --species=somespecies
Modify augustus.hints.gtf to point to the AUGUSTUS predictions with hints from previous BRAKER run; modify some_new_working_directory to the location where BRAKER should store results of the additional BRAKER run; this run will not modify AUGUSTUS parameters. We recommend that you specify the original species of the original run with --species=somespecies . Otherwise, BRAKER will create an unneeded species parameters directory Sp_* .
If --UTR=on is enabled, strand-separated bam-files can be provided with --bam=plus.bam,minus.bam . In that case, --stranded=... should hold the strands of the bam files ( + for plus strand, - for minus strand, . for unstranded). Note that unstranded data will be used in the gene prediction step, only, if the parameter --stranded=... is set. This is an experimental feature! GUSHR currently does not take advantage of stranded data.
If --makehub and [email protected] (with your valid e-mail adress) are provided, a track data hub for visualizing results with the UCSC Genome Browser will be generated using MakeHub (https://github.com/Gaius-Augustus/MakeHub).
By default, GeneMark-ES/ET/EP/ETP uses a probability of 0.001 for predicting the donor splice site pattern GC (instead of GT). It may make sense to increase this value for species where this donor splice site is more common. For example, in the species Emiliania huxleyi , about 50% of donor splice sites have the pattern GC (https://media.nature.com/original/nature-assets/nature/journal/v499/n7457/extref/nature12221-s2.pdf, page 5).
Use a species-specific lineage, eg arthropoda_odb10 for an arthropod. BRAKER does not support auto-typing of the lineage.
Specifying a BUSCO-lineage invokes two changes in BRAKER R28 :
BRAKER will run compleasm with the specified lineage in genome mode and convert the detected BUSCO matches into hints for AUGUSTUS. This may increase the number of BUSCOs in the augustus.hints.gtf file slightly.
BRAKER will invoke best_by_compleasm.py to check whether the braker.gtf file that is by default generated by TSEBRA has the lowest amount of missing BUSCOs compared to the augustus.hints.gtf and the genemark.gtf file. If not, the following decision schema is applied to re-run TSEBRA to minimize the missing BUSCOs in the final output of BRAKER (always braker.gtf). If an alternative and better gene set is created, the original braker.gtf gene set is moved to a directory called braker_original. Information on what happened during the best_by_compleasm.py run is written to the file best_by_compleasm.log.
![best_by_busco[fig14]](https://images.downcodes.com/uploads/20250214/img_67aee79a11fd439.png)
Please note that using BUSCO to assess the quality of a gene set, in particular when comparing BRAKER to other pipelines, does not make sense once you specified a BUSCO lineage. We recommend that you use other measures to assess the quality of your gene set, eg by comparing it to a reference gene set or running OMArk.
BRAKER produces several important output files in the working directory.
braker.gtf: Final gene set of BRAKER. This file may contain different contents depending on how you called BRAKER
in ETPmode: Final gene set of BRAKER consisting of genes predicted by AUGUSTUS and GeneMark-ETP that were combined and filtered by TSEBRA.
otherwise: Union of augustus.hints.gtf and reliable GeneMark-ES/ET/EP predictions (genes fully supported by external evidence). In --esmode , this is the union of augustus.ab_initio.gtf and all GeneMark-ES genes. Thus, this set is generally more sensitive (more genes correctly predicted) and can be less specific (more false-positive predictions can be present). This output is not necessarily better than augustus.hints.gtf, and it is not recommended to use it if BRAKER was run in ESmode.
braker.codingseq: Final gene set with coding sequences in FASTA format
braker.aa: Final gene set with protein sequences in FASTA format
braker.gff3: Final gene set in gff3 format (only produced if the flag --gff3 was specified to BRAKER.
Augustus/*: Augustus gene set(s) in as gtf/conding/aa files
GeneMark-E*/genemark.gtf: Genes predicted by GeneMark-ES/ET/EP/EP+/ETP in GTF-format.
hintsfile.gff: The extrinsic evidence data extracted from RNAseq.bam and/or protein data.
braker_original/*: Genes predicted by BRAKER (TSEBRA merge) before compleasm was used to improve BUSCO completeness
bbc/*: output folder of best_by_compleasm.py script from TSEBRA that is used to improve BUSCO completeness in the final output of BRAKER
Output files may be present with the following name endings and formats:
Coding sequences in FASTA-format are produced if the flag --skipGetAnnoFromFasta was not set.
Protein sequence files in FASTA-format are produced if the flag --skipGetAnnoFromFasta was not set.
For details about gtf format, see http://www.sanger.ac.uk/Software/formats/GFF/. A GTF-format file contains one line per predicted exon. Ejemplo:
HS04636 AUGUSTUS initial 966 1017 . + 0 transcript_id "g1.1"; gene_id "g1";
HS04636 AUGUSTUS internal 1818 1934 . + 2 transcript_id "g1.1"; gene_id "g1";
The columns (fields) contain:
seqname source feature start end score strand frame transcript ID and gene ID
If the --makehub option was used and MakeHub is available on your system, a hub directory beginning with the name hub_ will be created. Copy this directory to a publicly accessible web server. A file hub.txt resides in the directory. Provide the link to that file to the UCSC Genome Browser for visualizing results.
An incomplete example data set is contained in the directory BRAKER/example . In order to complete the data set, please download the RNA-Seq alignment file (134 MB) with wget :
cd BRAKER/example
wget http://topaz.gatech.edu/GeneMark/Braker/RNAseq.bam
In case you have trouble accessing that file, there's also a copy available from another server:
cd BRAKER/example
wget http://bioinf.uni-greifswald.de/augustus/datasets/RNAseq.bam
The example data set was not compiled in order to achieve optimal prediction accuracy, but in order to quickly test pipeline components. The small subset of the genome used in these test examples is not long enough for BRAKER training to work well.
Data corresponds to the last 1,000,000 nucleotides of Arabidopsis thaliana 's chromosome Chr5, split into 8 artificial contigs.
RNA-Seq alignments were obtained by VARUS.
The protein sequences are a subset of OrthoDB v10 plants proteins.
List of files:
genome.fa - genome file in fasta formatRNAseq.bam - RNA-Seq alignment file in bam format (this file is not a part of this repository, it must be downloaded separately from http://topaz.gatech.edu/GeneMark/Braker/RNAseq.bam)RNAseq.hints - RNA-Seq hints (can be used instead of RNAseq.bam as RNA-Seq input to BRAKER)proteins.fa - protein sequences in fasta formatThe below given commands assume that you configured all paths to tools by exporting bash variables or that you have the necessary tools in your $PATH.
The example data set also contains scripts tests/test*.sh that will execute below listed commands for testing BRAKER with the example data set. You find example results of AUGUSTUS and GeneMark-ES/ET/EP/ETP in the folder results/test* . Be aware that BRAKER contains several parts where random variables are used, ie results that you obtain when running the tests may not be exactly identical. To compare your test results with the reference ones, you can use the compare_intervals_exact.pl script as follows:
# Compare CDS features
compare_intervals_exact.pl --f1 augustus.hints.gtf --f2 ../../results/test${N}/augustus.hints.gtf --verbose
# Compare transcripts
compare_intervals_exact.pl --f1 augustus.hints.gtf --f2 ../../results/test${N}/augustus.hints.gtf --trans --verbose
Several tests use --gm_max_intergenic 10000 option to make the test runs faster. It is not recommended to use this option in real BRAKER runs, the speed increase achieved by adjusting this option is negligible on full-sized genomes.
We give runtime estimations derived from computing on Intel(R) Xeon(R) CPU E5530 @ 2.40GHz .
The following command will run the pipeline according to Figure 3:
braker.pl --genome genome.fa --bam RNAseq.bam --threads N --busco_lineage=lineage_odb10
This test is implemented in test1.sh , expected runtime is ~20 minutes.
The following command will run the pipeline according to Figure 4:
braker.pl --genome genome.fa --prot_seq proteins.fa --threads N --busco_lineage=lineage_odb10
This test is implemented in test2.sh , expected runtime is ~20 minutes.
The following command will run a pipeline that first trains GeneMark-ETP with protein and RNA-Seq hints and subsequently trains AUGUSTUS on the basis of GeneMark-ETP predictions. AUGUSTUS predictions are also performed with hints from both sources, see Figure 5.
Run with local RNA-Seq file:
braker.pl --genome genome.fa --prot_seq proteins.fa --bam ../RNAseq.bam --threads N --busco_lineage=lineage_odb10
This test is implemented in test3.sh , expected runtime is ~20 minutes.
Download RNA-Seq library from Sequence Read Archive (~1gb):
braker.pl --genome genome.fa --prot_seq proteins.fa --rnaseq_sets_ids ERR5767212 --threads N --busco_lineage=lineage_odb10
This test is implemented in test3_4.sh , expected runtime is ~35 minutes.
The training step of all pipelines can be skipped with the option --skipAllTraining . This means, only AUGUSTUS predictions will be performed, using pre-trained, already existing parameters. For example, you can predict genes with the command:
braker.pl --genome=genome.fa --bam RNAseq.bam --species=arabidopsis
--skipAllTraining --threads N
This test is implemented in test4.sh , expected runtime is ~1 minute.
The following command will run the pipeline with no extrinsic evidence:
braker.pl --genome=genome.fa --esmode --threads N
This test is implemented in test5.sh , expected runtime is ~20 minutes.
The following command will run BRAKER with training UTR parameters from RNA-Seq coverage data:
braker.pl --genome genome.fa --bam RNAseq.bam --UTR=on --threads N
This test is implemented in test6.sh , expected runtime is ~20 minutes.
The following command will add UTRs to augustus.hints.gtf from RNA-Seq coverage data:
braker.pl --genome genome.fa --bam RNAseq.bam --addUTR=on --threads N
This test is implemented in test7.sh , expected runtime is ~20 minutes.
There is currently no clean way to restart a failed BRAKER run (after solving some problem). However, it is possible to start a new BRAKER run based on results from a previous run -- given that the old run produced the required intermediate results. We will in the following refer to the old working directory with variable ${BRAKER_OLD} , and to the new BRAKER working directory with ${BRAKER_NEW} . The file what-to-cite.txt will always only refer to the software that was actually called by a particular run. You might have to combine the contents of ${BRAKER_NEW}/what-to-cite.txt with ${BRAKER_OLD}/what-to-cite.txt for preparing a publication. The following figure illustrates at which points BRAKER run may be intercepted.
![braker-intercept[fig8]](https://images.downcodes.com/uploads/20250214/img_67aee79a12cab310.png)
Figure 10: Points for intercepting a BRAKER run and reusing intermediate results in a new BRAKER run.
This option is only possible for BRAKER in ETmode or EPmode and no in ETPmode!
If you have access to an existing BRAKER output that contains hintsfiles that were generated from extrinsic data, such as RNA-Seq or protein sequences, you can recycle these hints files in a new BRAKER run. Also, hints from a separate ProtHint run can be directly used in BRAKER.
The hints can be given to BRAKER with --hints ${BRAKER_OLD}/hintsfile.gff option. This is illustrated in the test files test1_restart1.sh , test2_restart1.sh , test4_restart1.sh . The other modes (for which this test is missing) cannot be restarted in this way.
The GeneMark result can be given to BRAKER with --geneMarkGtf ${BRAKER_OLD}/GeneMark*/genemark.gtf option if BRAKER is run in ETmode or EPmode. This is illustrated in the test files test1_restart2.sh , test2_restart2.sh , test5_restart2.sh .
In ETPmode, you can either provide BRAKER with the results of the GeneMarkETP step manually, with --geneMarkGtf ${BRAKER_OLD}/GeneMark-ETP/proteins.fa/genemark.gtf , --traingenes ${BRAKER_OLD}/GeneMark-ETP/training.gtf , and --hints ${BRAKER_OLD}/hintsfile.gff (see test3_restart1.sh for an example), or you can specify the previous GeneMark-ETP results with the option --gmetp_results_dir ${BRAKER_OLD}/GeneMark-ETP/ so that BRAKER can search for the files automatically (see test3_restart2.sh for an example).
The trained species parameters for AGUSTUS can be passed with --skipAllTraining and --species $speciesName options. This is illustrated in test*_restart3.sh files. Note that in ETPmode you have to specify the GeneMark files as described in Option 2!
Before reporting bugs, please check that you are using the most recent versions of GeneMark-ES/ET/EP/ETP, AUGUSTUS and BRAKER. Also, check the list of Common problems, and the Issue list on GitHub before reporting bugs. We do monitor open issues on GitHub. Sometimes, we are unable to help you, immediately, but we try hard to solve your problems.
If you found a bug, please open an issue at https://github.com/Gaius-Augustus/BRAKER/issues (or contact [email protected] or [email protected]).
Information worth mentioning in your bug report:
Check in braker/yourSpecies/braker.log at which step braker.pl crashed.
There are a number of other files that might be of interest, depending on where in the pipeline the problem occurred. Some of the following files will not be present if they did not contain any errors.
braker/yourSpecies/errors/bam2hints.*.stderr - will give details on a bam2hints crash (step for converting bam file to intron gff file)
braker/yourSpecies/hintsfile.gff - is this file empty? If yes, something went wrong during hints generation - does this file contain hints from source “b2h” and of type “intron”? If not: GeneMark-ET will not be able to execute properly. Conversely, GeneMark-EP+ will not be able to execute correctly if hints from the source "ProtHint" are missing.
braker/yourSpecies/spaln/*err - errors reported by spaln
braker/yourSpecies/errors/GeneMark-{ET,EP,ETP}.stderr - errors reported by GeneMark-ET/EP+/ETP
braker/yourSpecies/errors/GeneMark-{ET,EP,ETP).stdout - may give clues about the point at which errors in GeneMark-ET/EP+/ETP occured
braker/yourSpecies/GeneMark-{ET,EP,ETP}/genemark.gtf - is this file empty? If yes, something went wrong during executing GeneMark-ET/EP+/ETP
braker/yourSpecies/GeneMark-{ET,EP}/genemark.f.good.gtf - is this file empty? If yes, something went wrong during filtering GeneMark-ET/EP+ genes for training AUGUSTUS
braker/yourSpecies/genbank.good.gb - try a “grep -c LOCUS genbank.good.gb” to determine the number of training genes for training AUGUSTUS, should not be low
braker/yourSpecies/errors/firstetraining.stderr - contains errors from first iteration of training AUGUSTUS
braker/yourSpecies/errors/secondetraining.stderr - contains errors from second iteration of training AUGUSTUS
braker/yourSpecies/errors/optimize_augustus.stderr - contains errors optimize_augustus.pl (additional training set for AUGUSTUS)
braker/yourSpecies/errors/augustus*.stderr - contain AUGUSTUS execution errors
braker/yourSpecies/startAlign.stderr - if you provided a protein fasta file, something went wrong during protein alignment
braker/yourSpecies/startAlign.stdout - may give clues on at which point protein alignment went wrong
BRAKER complains that the RNA-Seq file does not correspond to the provided genome file, but I am sure the files correspond to each other!
Please check the headers of the genome FASTA file. If the headers are long and contain whitespaces, some RNA-Seq alignment tools will truncate sequence names in the BAM file. This leads to an error with BRAKER. Solution: shorten/simplify FASTA headers in the genome file before running the RNA-Seq alignment and BRAKER.
GeneMark fails!
(a) GeneMark by default only uses contigs longer than 50k for training. If you have a highly fragmented assembly, this might lead to "no data" for training. You can override the default minimal length by setting the BRAKER argument --min_contig=10000 .
(b) see "[something] failed to execute" below.
[something] failed to execute!
When providing paths to software to BRAKER, please use absolute, non-abbreviated paths. For example, BRAKER might have problems with --SAMTOOLS_PATH=./samtools/ or --SAMTOOLS_PATH=~/samtools/ . Please use SAMTOOLS_PATH=/full/absolute/path/to/samtools/ , instead. This applies to all path specifications as command line options to braker.pl . Relative paths and absolute paths will not pose problems if you export a bash variable, instead, or if you append the location of tools to your $PATH variable.
GeneMark-ETP in BRAKER dies with '/scratch/11232323': No such file or directory.
This appears to be related to sorting large files, and it's a system configuration depending problem. Solve it with export TMPDIR=/tmp/ before calling BRAKER via Singularity.
BRAKER cannot find the Augustus script XYZ...
Update Augustus from github with git clone https://github.com/Gaius-Augustus/Augustus.git . Do not use Augustus from other sources. BRAKER is highly dependent on an up-to-date Augustus. Augustus releases happen rather rarely, updates to the Augustus scripts folder occur rather frequently.
Does BRAKER depend on Python3?
Lo hace. The python scripts employed by BRAKER are not compatible with Python2.
Why does BRAKER predict more genes than I expected?
If transposable elements (or similar) have not been masked appropriately, AUGUSTUS tends to predict those elements as protein coding genes. This can lead to a huge number genes. You can check whether this is the case for your project by BLASTing (or DIAMONDing) the predicted protein sequences against themselves (all vs. all) and counting how many of the proteins have a high number of high quality matches. You can use the output of this analysis to divide your gene set into two groups: the protein coding genes that you want to find and the repetitive elements that were additionally predicted.
I am running BRAKER in Anaconda and something fails...
Update AUGUSTUS and BRAKER from github with git clone https://github.com/Gaius-Augustus/Augustus.git and git clone https://github.com/Gaius-Augustus/BRAKER.git . The Anaconda installation is great, but it relies on releases of AUGUSTUS and BRAKER - which are often lagging behind. Please use the current GitHub code, instead.
Why and where is the GenomeThreader support gone?
BRAKER is a joint project between teams from University of Greifswald and Georgia Tech. While the group of Mark Bordovsky from Georgia Tech contributes GeneMark expertise, the group of Mario Stanke from University of Greifswald contributes AUGUSTUS expertise. Using GenomeThreader to build training genes for AUGUSTUS in BRAKER circumvents execution of GeneMark. Thus, the GenomeThreader mode is strictly speaking not part of the BRAKER project. The previous functionality of BRAKER with GenomeThreader has been moved to GALBA at https://github.com/Gaius-Augustus/GALBA. Note that GALBA has also undergone extension for using Miniprot instead of GenomeThreader.
My BRAKER gene set has too many BUSCO duplicates!
AUGUSTUS within BRAKER can predict alternative splicing isoforms. Also the merge of the AUGUSTUS and GeneMark gene set by TSEBRA within BRAKER may result in additional isoforms for a single gene. The BUSCO duplicates usually come from alternative splicing isoforms, ie they are expected.
Augustus and/or etraining within BRAKER complain that the file aug_cmdln_parameters.json is missing. Even though I am using the latest Singularity container!
BRAKER copies the AUGUSTUS_CONFIG_PATH folder to a writable location. In older versions of Augustus, that file was indeed not existing. If the local writable copy of a folder already exists, BRAKER will not re-copy it. Simply delete the old folder. (It is often ~/.augustus , so you can simply do rm -rf ~/.augustus ; the folder might be residing in $PWD if your home directory was not writable).
I sit behind a firewall, compleasm cannot download the BUSCO files, what can I do? See Issue #785 (comment)
Since BRAKER is a pipeline that calls several Bioinformatics tools, publication of results obtained by BRAKER requires that not only BRAKER is cited, but also the tools that are called by BRAKER. BRAKER will output a file what-to-cite.txt in the BRAKER working directory, informing you about which exact sources apply to your run.
Always cite:
Stanke, M., Diekhans, M., Baertsch, R. and Haussler, D. (2008). Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics, doi: 10.1093/bioinformatics/btn013.
Stanke. M., Schöffmann, O., Morgenstern, B. and Waack, S. (2006). Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics 7, 62.
If you provided any kind of evidence for BRAKER, cite:
If you provided both short read RNA-Seq evidence and a large database of proteins, cite:
Gabriel, L., Bruna, T., Hoff, KJ, Ebel, M., Lomsadze, A., Borodovsky, M., Stanke, M. (2023). BRAKER3: Fully Automated Genome Annotation Using RNA-Seq and Protein Evidence with GeneMark-ETP, AUGUSTUS and TSEBRA. bioRxiV, doi: 10.1101/2023.06.10.54444910.1101/2023.01.01.474747.
Bruna, T., Lomsadze, A., Borodovsky, M. (2023). GeneMark-ETP: Automatic Gene Finding in Eukaryotic Genomes in Consistence with Extrinsic Data. bioRxiv, doi: 10.1101/2023.01.13.524024.
Kovaka, S., Zimin, AV, Pertea, GM, Razaghi, R., Salzberg, SL, & Pertea, M. (2019). Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome biology, 20(1):1-13.
Pertea, G., & Pertea, M. (2020). GFF utilities: GffRead and GffCompare. F1000Research, 9.
Quinlan, AR (2014). BEDTools: the Swiss‐army tool for genome feature analysis. Current protocols in bioinformatics, 47(1):11-12.
If the only source of evidence for BRAKER was a large database of protein sequences, cite:
If the only source of evidence for BRAKER was RNA-Seq data, cite:
Hoff, KJ, Lange, S., Lomsadze, A., Borodovsky, M. and Stanke, M. (2016). BRAKER1: unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics, 32(5):767-769.
Lomsadze, A., Paul DB, and Mark B. (2014) Integration of Mapped Rna-Seq Reads into Automatic Training of Eukaryotic Gene Finding Algorithm. Nucleic Acids Research 42(15): e119--e119
If you called BRAKER3 with an IsoSeq BAM file, or if you envoked the --busco_lineage option, cite:
If you called BRAKER with the --busco_lineage option, in addition, cite:
Simão, FA, Waterhouse, RM, Ioannidis, P., Kriventseva, EV, & Zdobnov, EM (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics, 31(19), 3210-3212.
Li, H. (2023). Protein-to-genome alignment with miniprot. Bioinformatics, 39(1), btad014.
Huang, N., & Li, H. (2023). compleasm: a faster and more accurate reimplementation of BUSCO. Bioinformatics, 39(10), btad595.
If any kind of AUGUSTUS training was performed by BRAKER, check carefully whether you configured BRAKER to use NCBI BLAST or DIAMOND. One of them was used to filter out redundant training gene structures.
If you used NCBI BLAST, please cite:
Altschul, AF, Gish, W., Miller, W., Myers, EW and Lipman, DJ (1990). A basic local alignment search tool. J Mol Biol 215:403--410.
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., and Madden, TL (2009). Blast+: architecture and applications. BMC bioinformatics, 10(1):421.
If you used DIAMOND, please cite:
If BRAKER was executed with a genome file and no extrinsic evidence, cite, then GeneMark-ES was used, cite:
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, YO and Borodovsky, M. (2005). Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Research, 33(20):6494--6506.
Ter-Hovhannisyan, V., Lomsadze, A., Chernoff, YO and Borodovsky, M. (2008). Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome research, pages gr--081612, 2008.
Hoff, KJ, Lomsadze, A., Borodovsky, M. and Stanke, M. (2019). Whole-Genome Annotation with BRAKER. Methods Mol Biol. 1962:65-95, doi: 10.1007/978-1-4939-9173-0_5.
If BRAKER was run with proteins as source of evidence, please cite all tools that are used by the ProtHint pipeline to generate hints:
Bruna, T., Lomsadze, A., & Borodovsky, M. (2020). GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genomics and Bioinformatics, 2(2), lqaa026.
Buchfink, B., Xie, C., Huson, DH (2015). Fast and sensitive protein alignment using DIAMOND. Nature Methods 12:59-60.
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, YO and Borodovsky, M. (2005). Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Research, 33(20):6494--6506.
Iwata, H., and Gotoh, O. (2012). Benchmarking spliced alignment programs including Spaln2, an extended version of Spaln that incorporates additional species-specific features. Nucleic acids research, 40(20), e161-e161.
Gotoh, O., Morita, M., Nelson, DR (2014). Assessment and refinement of eukaryotic gene structure prediction with gene-structure-aware multiple protein sequence alignment. BMC bioinformatics, 15(1), 189.
If BRAKER was executed with RNA-Seq alignments in bam-format, then SAMtools was used, cite:
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R.; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25(16):2078-9.
Barnett, DW, Garrison, EK, Quinlan, AR, Strömberg, MP and Marth GT (2011). BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics, 27(12):1691-2
If BRAKER downloaded RNA-Seq libraries from SRA using their IDs, cite SRA, SRA toolkit, and HISAT2:
Leinonen, R., Sugawara, H., Shumway, M., & International Nucleotide Sequence Database Collaboration. (2010). The sequence read archive. Nucleic acids research, 39(suppl_1), D19-D21.
SRA Toolkit Development Team (2020). SRA Toolkit. https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software.
Kim, D., Paggi, JM, Park, C., Bennett, C., & Salzberg, SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature biotechnology, 37(8):907-915.
If BRAKER was executed using RNA-Seq data in FASTQ format, cite HISAT2:
If BRAKER called MakeHub for creating a track data hub for visualization of BRAKER results with the UCSC Genome Browser, cite:
If BRAKER called GUSHR for generating UTRs, cite:
Keilwagen, J., Hartung, F., Grau, J. (2019) GeMoMa: Homology-based gene prediction utilizing intron position conservation and RNA-seq data. Methods Mol Biol. 1962:161-177, doi: 10.1007/978-1-4939-9173-0_9.
Keilwagen, J., Wenk, M., Erickson, JL, Schattat, MH, Grau, J., Hartung F. (2016) Using intron position conservation for homology-based gene prediction. Nucleic Acids Research, 44(9):e89.
Keilwagen, J., Hartung, F., Paulini, M., Twardziok, SO, Grau, J. (2018) Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinformatics, 19(1):189.
All source code, ie scripts/*.pl or scripts/*.py are under the Artistic License (see http://www.opensource.org/licenses/artistic-license.php).
[F1] EX = ES/ET/EP/ETP, all available for download under the name GeneMark-ES/ET/EP ↩
[F2] Please use the latest version from the master branch of AUGUSTUS distributed by the original developers, it is available from github at https://github.com/Gaius-Augustus/Augustus. Problems have been reported from users that tried to run BRAKER with AUGUSTUS releases maintained by third parties, ie Bioconda. ↩
[F4] install with sudo apt-get install cpanminus ↩
[F6] The binary may eg reside in bamtools/build/src/toolkit ↩
[R0] Bruna, Tomas, Hoff, Katharina J., Lomsadze, Alexandre, Stanke, Mario, and Borodovsky, Mark. 2021. “BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database." NAR Genomics and Bioinformatics 3(1):lqaa108.↩
[R1] Hoff, Katharina J, Simone Lange, Alexandre Lomsadze, Mark Borodovsky, and Mario Stanke. 2015. “BRAKER1: Unsupervised Rna-Seq-Based Genome Annotation with Genemark-et and Augustus.” Bioinformatics 32 (5). Oxford University Press: 767--69.↩
[R2] Lomsadze, Alexandre, Paul D Burns, and Mark Borodovsky. 2014. “Integration of Mapped Rna-Seq Reads into Automatic Training of Eukaryotic Gene Finding Algorithm.” Nucleic Acids Research 42 (15). Oxford University Press: e119--e119.↩
[R3] Stanke, Mario, Mark Diekhans, Robert Baertsch, and David Haussler. 2008. “Using Native and Syntenically Mapped cDNA Alignments to Improve de Novo Gene Finding.” Bioinformatics 24 (5). Oxford University Press: 637--44.↩
[R4] Stanke, Mario, Oliver Schöffmann, Burkhard Morgenstern, and Stephan Waack. 2006. “Gene Prediction in Eukaryotes with a Generalized Hidden Markov Model That Uses Hints from External Sources.” BMC Bioinformatics 7 (1). BioMed Central: 62.↩
[R5] Barnett, Derek W, Erik K Garrison, Aaron R Quinlan, Michael P Strömberg, and Gabor T Marth. 2011. “BamTools: A C++ Api and Toolkit for Analyzing and Managing Bam Files.” Bioinformatics 27 (12). Oxford University Press: 1691--2.↩
[R6] Li, Heng, Handsaker, Bob, Alec Wysoker, Tim Fennell, Jue Ruan, Nils Homer, Gabor Marth, Goncalo Abecasis, and Richard Durbin. 2009. “The Sequence Alignment/Map Format and Samtools.” Bioinformatics 25 (16). Oxford University Press: 2078--9.↩
[R7] Gremme, G. 2013. “Computational Gene Structure Prediction.” PhD thesis, Universität Hamburg.↩
[R8] Gotoh, Osamu. 2008a. “A Space-Efficient and Accurate Method for Mapping and Aligning cDNA Sequences onto Genomic Sequence.” Nucleic Acids Research 36 (8). Oxford University Press: 2630--8.↩
[R9] Iwata, Hiroaki, and Osamu Gotoh. 2012. “Benchmarking Spliced Alignment Programs Including Spaln2, an Extended Version of Spaln That Incorporates Additional Species-Specific Features.” Nucleic Acids Research 40 (20). Oxford University Press: e161--e161.↩
[R10] Osamu Gotoh. 2008b. “Direct Mapping and Alignment of Protein Sequences onto Genomic Sequence.” Bioinformatics 24 (21). Oxford University Press: 2438--44.↩
[R11] Slater, Guy St C, and Ewan Birney. 2005. “Automated Generation of Heuristics for Biological Sequence Comparison.” BMC Bioinformatics 6(1). BioMed Central: 31.↩
[R12] Altschul, SF, W. Gish, W. Miller, EW Myers, and DJ Lipman. 1990. “Basic Local Alignment Search Tool.” Journal of Molecular Biology 215:403--10.↩
[R13] Camacho, Christiam, et al. 2009. “BLAST+: architecture and applications.“ BMC Bioinformatics 1(1): 421.↩
[R14] Lomsadze, A., V. Ter-Hovhannisyan, YO Chernoff, and M. Borodovsky. 2005. “Gene identification in novel eukaryotic genomes by self-training algorithm.” Nucleic Acids Research 33 (20): 6494--6506. doi:10.1093/nar/gki937.↩
[R15] Ter-Hovhannisyan, Vardges, Alexandre Lomsadze, Yury O Chernoff, and Mark Borodovsky. 2008. “Gene Prediction in Novel Fungal Genomes Using an Ab Initio Algorithm with Unsupervised Training.” Genome Research . Cold Spring Harbor Lab, gr--081612.↩
[R16] Hoff, KJ 2019. MakeHub: Fully automated generation of UCSC Genome Browser Assembly Hubs. Genomics, Proteomics and Bioinformatics , in press, preprint on bioarXive, doi: https://doi.org/10.1101/550145.↩
[R17] Bruna, T., Lomsadze, A., & Borodovsky, M. 2020. GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genomics and Bioinformatics, 2(2), lqaa026. doi: https://doi.org/10.1093/nargab/lqaa026.↩
[R18] Kriventseva, EV, Kuznetsov, D., Tegenfeldt, F., Manni, M., Dias, R., Simão, FA, and Zdobnov, EM 2019. OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Research, 47(D1), D807-D811.↩
[R19] Keilwagen, J., Hartung, F., Grau, J. (2019) GeMoMa: Homology-based gene prediction utilizing intron position conservation and RNA-seq data. Methods Mol Biol. 1962:161-177, doi: 10.1007/978-1-4939-9173-0_9.↩
[R20] Keilwagen, J., Wenk, M., Erickson, JL, Schattat, MH, Grau, J., Hartung F. (2016) Using intron position conservation for homology-based gene prediction. Nucleic Acids Research, 44(9):e89.↩
[R21] Keilwagen, J., Hartung, F., Paulini, M., Twardziok, SO, Grau, J. (2018) Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinformatics, 19(1):189.↩
[R22] SRA Toolkit Development Team (2020). SRA Toolkit. https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software.[↩](#a22)
[R23] Kim, D., Paggi, JM, Park, C., Bennett, C., & Salzberg, SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature biotechnology, 37(8):907-915.↩
[R24] Quinlan, AR (2014). BEDTools: the Swiss‐army tool for genome feature analysis. Current protocols in bioinformatics, 47(1):11-12.↩
[R25] Kovaka, S., Zimin, AV, Pertea, GM, Razaghi, R., Salzberg, SL, & Pertea, M. (2019). Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome biology, 20(1):1-13.↩
[R26] Pertea, G., & Pertea, M. (2020). GFF utilities: GffRead and GffCompare. F1000Research, 9.↩
[R27] Huang, N., & Li, H. (2023). compleasm: a faster and more accurate reimplementation of BUSCO. Bioinformatics, 39(10), btad595.↩
[R28] Bruna, T., Gabriel, L. & Hoff, KJ (2024). Navigating Eukaryotic Genome Annotation Pipelines: A Route Map to BRAKER, Galba, and TSEBRA. arXiv, https://doi.org/10.48550/arXiv.2403.19416 .↩


