SCDC
1.0.0
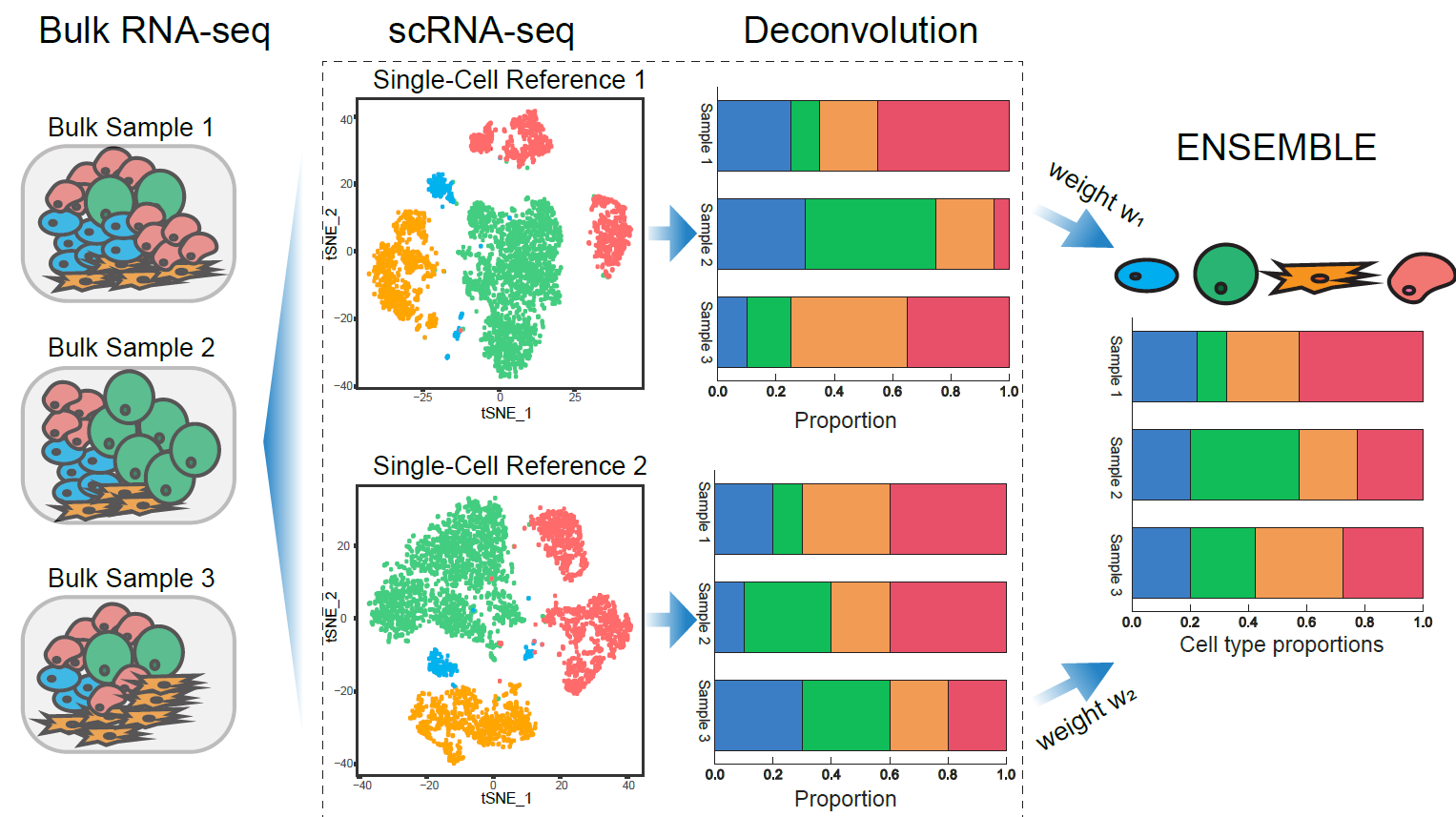
SCDC es un método de deconvolución para ARN-seq a granel que aprovecha las expresiones genéticas específicas de tipo celular de múltiples conjuntos de datos de referencia SCRNA-Seq . SCDC adopta un método de conjunto para integrar los resultados de la deconvolución de diferentes conjuntos de datos SCRNA-seq que se producen en diferentes laboratorios y en diferentes momentos, abordando implícitamente la confusión de efectos por lotes.
Meichen Dong, Aatish Thennavan, Eugene Urrutia, Yun Li, Charles M Perou, Fei Zou, Yuchao Jiang, SCDC: deconvolución de la expresión génica a granel por múltiples referencias de secuenciación de ARN de una sola célula, informes en bioinformática, BBZ166, https: // doi. org/10.1093/bib/bbz166
Licencia: MIT
Puede instalar la versión lanzada de SCDC desde GitHub con:
if (! require ("devTools")) {
install.packages ("DevTools")
} devTools :: install_github ("Meichendong/scdc")El problema del paquete de dependencia sobre 'XBIOC' podría resolverse mediante:
install.packages ("Remotes") Remotes :: install_github ("Renozao/XBIOC")Consulte la página Vignettes.
El documento SCDC se publica en sesiones informativas en bioinformática.
Las preguntas sobre el paquete se pueden enviar un correo electrónico a: [email protected]
Cuando solo hay 'un sujeto/individuo' en el conjunto de datos de una sola celda, use las funciones SCDC_qc_ONE() , SCDC_prop_ONE() .
Aspectos que podrían afectar los resultados de la deconvolución:
Formato de datos: ¿Son las muestras de celda a granel y un solo recuento sin procesar / mismo formato? Esperamos que el formato de datos sea consistente y comparable.
Filtrado de genes: ¿Filtró genes / genes ribosómicos / genes mitocondriales expresados de baja forma? Estos genes pueden afectar el análisis posterior.
Tamaño de celda y factores de tamaño de la biblioteca: para una sola celda, ¿cree que la suma de todos los recuentos de genes (el tamaño de la biblioteca) podría reflejar su tamaño de celda real? Este es uno de nuestros supuestos: la relación de los tamaños de biblioteca entre los tipos de células puede reflejar la relación de los tamaños de células reales entre los tipos de células. Si no, puede ingresar manualmente el factor de tamaño de la celda al construir la "matriz básica".
Tipos de células similares: ¿existen tipos de células que puedan confundir el análisis? Por ejemplo, tipos de células que tienen genes perfiles /marcadores muy similares.
Falta los tipos de células principales / problemas técnicos: ¿espera que el procedimiento de secuenciación marque una gran diferencia en el volumen y SC incluso la técnica es la misma? A veces, los datos de referencia de una sola celda pueden perder información para algunos tipos de células. Por ejemplo, hay células grasas en sus muestras a granel, pero de alguna manera no lo tiene para los datos de una sola celda.
Desconvolución utilizando un conjunto de datos de referencia único: ¿Intentó usar un conjunto de datos de referencia para probar si los resultados tienen sentido en general? Veo que probaste Bisque. ¿Has probado otros métodos como cibersortx? Si los resultados de otros métodos de deconvolución de "una referencia" tienen más sentido, entonces puede ingresarlos directamente usando nuestro paso de conjunto.


