ClockstaR
1.0.0

Sebastian Duchene, Martyna Molak et Simon YW Ho.
Laboratoire d'écologie moléculaire, évolution et phylogénétique (MEEP)
École des sciences biologiques
Université de Sydney
10 juin 2015
partition_data_partitionfinder('drag fasta file with concatenated data here', 'drag partition finder output here')
optim.trees.interactive(folder.parts = 'path to your folder with fasta files and tree topology here')
Implémenter l'optimisation des distances des arbres en utilisant la dérivée de la distance BSD
Implémenter la version parallèle pour la distance topologique
Rédiger un didacticiel sur le clustering de distance topologique
Intégrer le générateur de modèles pour les tests de modèles
Intégrez RaxML pour une optimisation maximale de la vraisemblance des longueurs et des topologies des branches
L'estimation des échelles de temps évolutives avec des ensembles de données multigéniques est un exercice courant dans les études phylogénétiques. Les ensembles de données multigéniques peuvent être divisés par gène, par position de codon ou par les deux. Dans ce didacticiel, nous faisons référence aux « sous-ensembles de données » comme à des gènes individuels ou à toute sous-unité de l'ensemble de données multigéniques. Le terme « partitions » fera référence à un groupe de sous-ensembles de données.
Bien que les sous-ensembles de données puissent être concaténés et analysés avec un seul modèle d'horloge détendu, les modèles de variation de taux entre lignées peuvent différer entre les sous-ensembles de données, même lorsque leurs topologies arborescentes sont identiques. Par exemple, la variation du taux de gènes mitochondriaux au sein d’une lignée peut différer de celle des gènes nucléaires. Par conséquent, différents modèles d'horloge relâchée peuvent être attribués à différents sous-ensembles de données afin d'améliorer les estimations des échelles de temps évolutives et l'ajustement statistique (voir Duchene et Ho., 2014a).
Il existe un grand nombre de façons de partitionner des ensembles de données multigènes. Une approche courante pour comparer les schémas de partitionnement consiste à utiliser des facteurs de Bayes ou des critères basés sur la vraisemblance pour l'ajustement du modèle. Dans la plupart des cas, cependant, il est impossible de tester tous les schémas de partitionnement possibles, en particulier avec des méthodes de calcul de facteurs Bayes à forte intensité de calcul.
ClockstaR estime la longueur des branches phylogénétiques de chaque sous-ensemble de données. La distance branche-score, connue sous le nom de sBSDmin, est calculée pour chaque paire d'arbres comme mesure de la différence dans leurs modèles de variation de taux entre lignées. Ces distances sont utilisées pour déduire la meilleure stratégie de partitionnement en utilisant la statistique GAP avec l'algorithme de clustering PAM, tel qu'implémenté dans le cluster de packages (Maechler et al., 2012) (pour plus de détails sur la métrique sBSDmin, voir Duchene et al., 2014b). .
ClockstaR est un package R pour les analyses d'horloge moléculaire phylogénétique d'ensembles de données multigéniques. Il utilise les modèles de variation du taux de lignée parmi les différents gènes pour sélectionner la stratégie de partitionnement d'horloge. La méthode utilise une métrique de distance d'arbre phylogénétique et un algorithme d'apprentissage automatique non supervisé pour identifier le nombre optimal de partitions d'horloge et quels gènes doivent être analysés sous chacune des partitions. La stratégie de partitionnement sélectionnée dans ClocsktaR peut être utilisée pour une analyse ultérieure de l'horloge moléculaire avec des programmes tels que BEAST, MrBayes, PhyloBayes et autres.
Veuillez suivre ce lien pour la publication originale.
ClockstaR nécessite une installation R. Cela nécessite également certaines dépendances R, qui peuvent être obtenues via R, comme expliqué ci-dessous.
Veuillez envoyer toute demande ou question à Sebastian Duchene (sebastian.duchene[at]sydney.edu.au). D'autres logiciels et ressources peuvent être trouvés au Laboratoire d'écologie moléculaire, d'évolution et de phylogénétique de l'Université de Sydney.
Téléchargez ce référentiel sous forme de fichier zip et décompressez-le. Les instructions suivantes utilisent le dossier clockstar_example_data, qui contient des fichiers fasta et un arbre phylogénétique au format newick. Ouvrez l'un de ces fichiers dans un éditeur de texte, tel que Text Wrangler. Ces données ont été simulées selon quatre modèles de variation du taux d'évolution. Notez que l'arborescence est la topologie arborescente de tous les gènes ou partitions de données. Pour exécuter ClockstaR, veuillez formater vos données de la même manière que les exemples de données dans clockstar_example_data.
ClockstaR peut être installé directement depuis GitHub. Cela nécessite le package devtools. Tapez le code suivant à l'invite R pour installer tous les outils nécessaires (notez que vous aurez besoin d'une connexion Internet pour télécharger directement les packages) :.
install . packages ( " devtools " )
library (devtools)
install_github ( ' ClockstaR ' , ' sebastianduchene ' )Après le téléchargement et l'installation, chargez ClockstaR avec la bibliothèque de fonctions .
library (ClockstaR2)Pour voir un exemple sur la façon dont le programme est exécuté, tapez :
example (ClockstaR2)Le reste de ce tutoriel utilise le dossier clockstar_example_data
La première étape consiste à obtenir les arbres génétiques pour chacun des alignements. Pour ce faire, nous utilisons la topologie arborescente et optimisons les longueurs de branche en utilisant chacun des alignements de gènes individuels, dans ce cas A1.fasta à C3.fasta. Si vous disposez des arbres génétiques, enregistrez-les au format Newick dans un fichier et passez à l'étape suivante (exécuter Clockstar de manière interactive).
Tapez le code suivant dans l'invite R et appuyez sur Entrée :
optim . trees . interactive ()Si vous recevez un message d'erreur concernant l'installation du package phangorn, veuillez utiliser ce code puis répétez optim.trees.interactive()
install . packcages ( " phangorn " )ClockstaR imprimera le message suivant :
Please drag a folder with the data subsets and a tree topology . The files should be in FASTA format, and the trees in NEWICKFaites glisser le dossier clockstar_example_data vers la console R et tapez Entrée. A noter que le dossier ne doit contenir que les alignements au format FASTA, et la topologie arborescente en NEWICK. Vous verrez le message suivant :
What should be the name of the file to save the optimised trees ?Tapez le nom du fichier pour les arbres optimisés. Dans ce cas nous utiliserons "exemple.trees"
example . treesÀ ce stade, ClockstaR demandera s’il doit utiliser un modèle de substitution distinct pour chaque gène ou utiliser JC dans tous les cas. Puisque ces données ont été simulées sous le JC, nous allons taper "n" et appuyer sur Entrée. Tapez « y » pour spécifier chaque modèle de substitution séparément.
Après avoir tapé « n » et appuyé sur Entrée, ClockstaR commencera à fonctionner. Il imprimera les arbres génétiques dans le périphérique graphique. Si l'arborescence spécifiée était enracinée, elle peut également afficher quelques avertissements, qui peuvent être ignorés en toute sécurité.
Ouvrez le dossier clockstar_example_data. Vous trouverez un fichier portant le nom "exemple.trees", comme spécifié quelques étapes ci-dessus. Ouvrez example.trees dans un éditeur de texte. Il contient chaque arbre génétique et les noms des arbres, correspondant aux noms des alignements de gènes. Cela devrait ressembler à ceci :
A1 . fasta (( t1 : 0.01504695462 ,( t2 : 0.00987 ...
A2 . fasta (( t1 : 0.01520523401 ,( t2 : 0.01317 ...
A3 . fasta (( t1 : 0.01519309467 ,( t2 : 0.01092 ...
.
.
.Ce fichier avec les arbres sera utilisé pour la prochaine étape.
Pour cette étape il est nécessaire de disposer des arbres génétiques dans un fichier, tel que celui obtenu à l’étape précédente.
Ouvrez R et chargez ClockstaR comme indiqué ci-dessus. Tapez le code suivant à l'invite :
clockstar . interactive ()ClockstaR imprimera le message suivant :
please drag or type in the path to your gene trees file in NEWICK format :Faites glisser le fichier contenant les arbres génétiques vers la console R. Si vous avez suivi l'étape précédente, le fichier s'appellera exemple.trees. Tapez Entrée.
En fonction des packages que vous avez installés, ClockstaR peut vous demander s'il doit s'exécuter en parallèle. Ceci est efficace pour les grands ensembles de données. Mais pour les données d'exemple, cela ne fera pas une grande différence, alors tapez "n" si vous voyez ce message, puis tapez enter :
Packages foreach and doParallel are available for parallel computation
Should we run ClockstaR in parallel (y / n) ? (This is good for large data sets)Clockstar va maintenant commencer à fonctionner. Le résultat à l'écran devrait ressembler à ceci :
[ 1 ] " Calculating sBSDmin distances between all pairs of trees "
[ 1 ] " Estimating tree distances "
[ 1 ] " estimating distances 1 of 11 "
[ 1 ] " estimating distances 2 of 11 "
[ 1 ] " estimating distances 3 of 11 "
[ 1 ] " estimating distances 4 of 11 "
[ 1 ] " estimating distances 5 of 11 "
.
.
.Après avoir estimé les distances des arbres (décrites dans la publication originale), ClockstaR imprimera le message suivant :
" I finished calculating the sBSDmin distances between trees "
The settings for clustering with ClockstaR are :
PAM clustering algorithm
K from 1 to number of data subsets - 1
SEmax criterion to select the optimal k
500 bootstrap replicates
Are these correct ? (y / n)Ce sont les paramètres de l'algorithme de clustering. Ils conviennent à la plupart des ensembles de données, donc dans cet exemple, nous pouvons taper « y » puis saisir. En tapant « n », nous pouvons modifier ces paramètres, pour plus de détails, voir Kaufman et Rousseeuw (2009).
ClockstaR va maintenant exécuter l'algorithme de clustering. À la fin, il imprimera le meilleur nombre de partitions et demandera si les résultats doivent être enregistrés dans un fichier pdf :
[ 1 ] " ClockstaR has finished running "
[ 1 ] " The best number of partitions for your data set is: 3 "
Do you wish to save the results in a pdf file ? (y / n)Tapez "y" puis entrez.
ClockstaR demandera alors le nom des fichiers de sortie :
What should be the name and path of the output file ?Pour cet exemple, tapez "example_run" et entrez, mais n'importe quel nom peut être utilisé.
Ouvrez maintenant le dossier clockstar_example_data et ouvrez les deux fichiers pdf, example_run_gapstats.pdf et example_run_matrix.pdf.
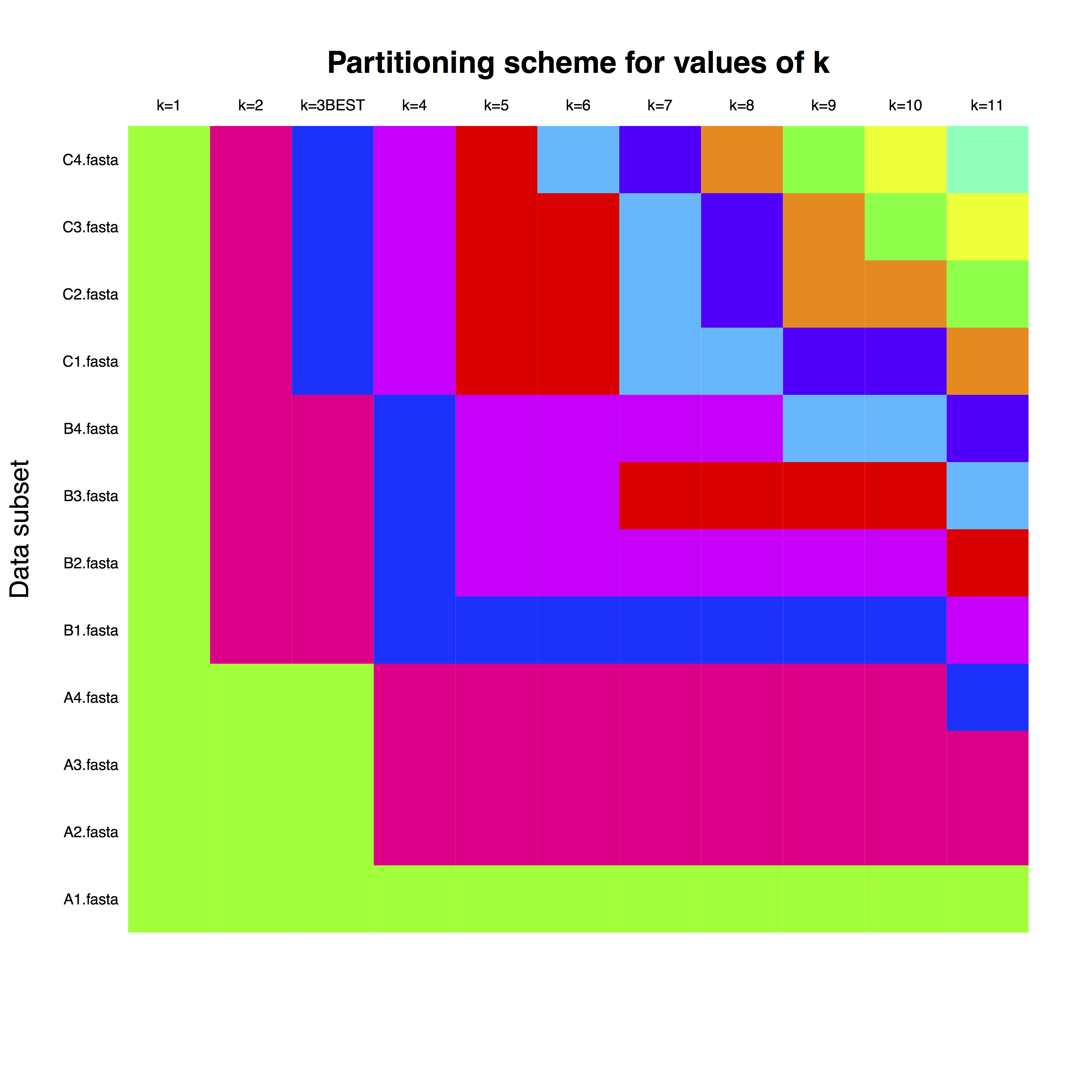
example_run_matrix est une matrice, où les lignes correspondent à chaque gène, comme nommé dans les fichiers FASTA. Les colonnes indiquent le nombre de partitions et les couleurs représentent l'affectation de chaque gène à la partition d'horloge. Par exemple, pour k = 3, qui est le meilleur nombre de partitions, on peut utiliser des partitions d'horloge distinctes pour les gènes portant les lettres A, B et C.
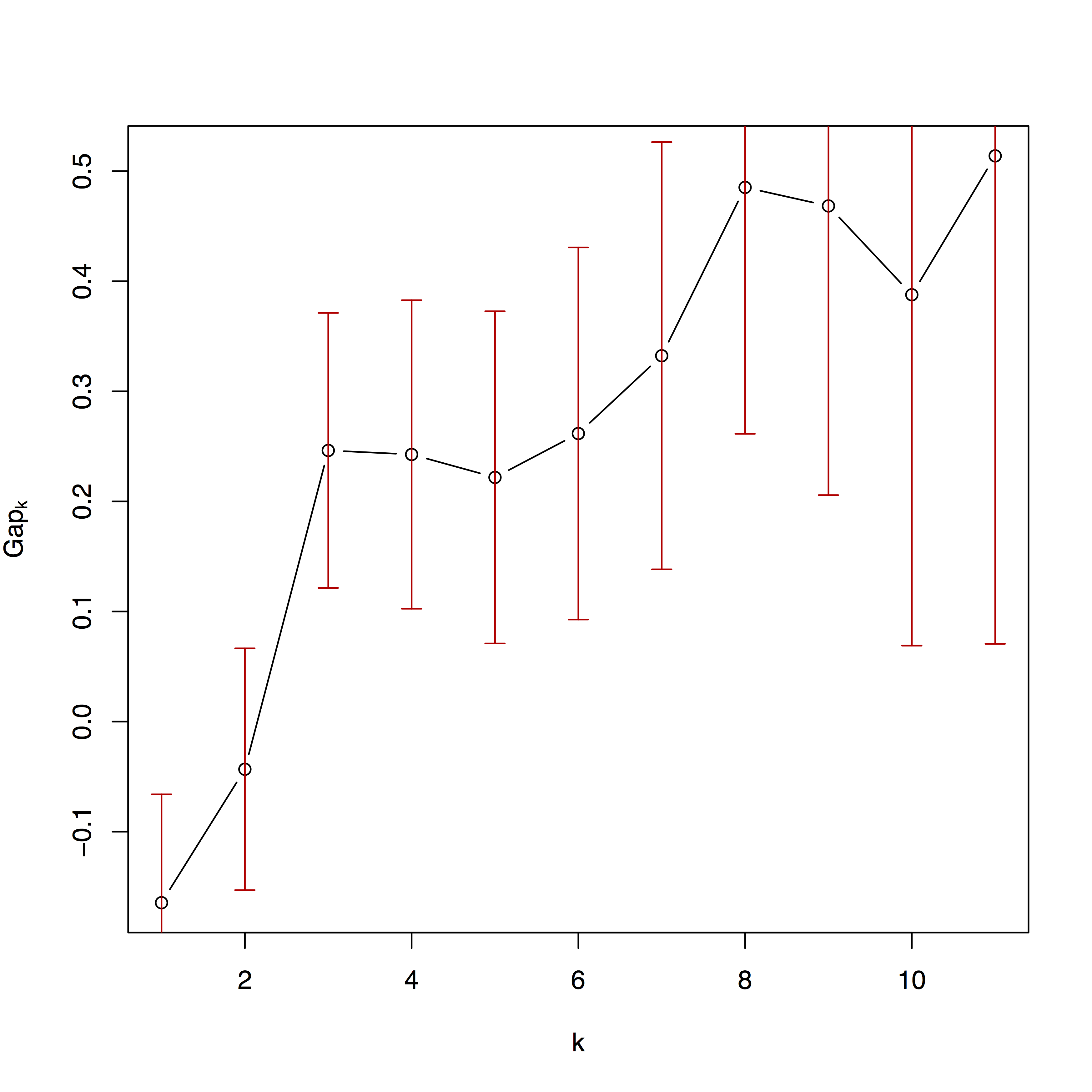
Le deuxième graphique est l’ajustement des algorithmes de clustering sur différents nombres de partitions. Plus de détails sont disponibles dans Kaufman et Rousseeuw (2009) et dans la documentation du cluster de packages.
ClockstaR peut être exécuté avec d'autres paramètres personnalisés. Veuillez consulter la documentation pour d'autres détails ou envoyez-moi un message pour toute question à sebastian.duchene[at]sydney.edy.au.
Le logo a été conçu par Jun Tong
Duchêne, S. et Ho, SY (2014a). Utilisation de plusieurs modèles d'horloge relâchés pour estimer les échelles de temps évolutives à partir des données de séquence d'ADN. Phylogénétique moléculaire et évolution (77) : 65-70.
Duchene, S., Molak, M. et Ho, SY (2014b). ClockstaR : choix du nombre de modèles d'horloge relâchée en analyse phylogénétique moléculaire. Bioinformatique 30 (7) : 1017-1019.
Kaufman, L. et Rousseeuw, PJ (2009). Recherche de groupes dans les données : une introduction à l'analyse groupée (Vol. 344). John Wiley et fils.


