Ce dépôt contient :
sepal nécessite python3 , de préférence une version ultérieure ou égale à 3.5. Pour télécharger et installer, ouvrez le terminal et accédez au répertoire dans lequel vous souhaitez que sepal soit téléchargé et faites :
git clone https://github.com/almaan/sepal.git
cd sepal
chmod +x setup.py
./setup.py install
En fonction de vos privilèges utilisateur, vous devrez peut-être ajouter --user comme argument à setup.py . L'exécution de l'installation vous donnera l'installation minimale requise pour calculer les temps de diffusion. Cependant, si vous souhaitez pouvoir utiliser les modules d'analyse, vous devez également installer les packages recommandés. Pour ce faire, exécutez simplement (dans le même répertoire) :
pip install -e " .[full] " encore une fois, le --user pourrait être nécessaire d'inclure. De plus, vous devrez peut-être utiliser pip3 si c'est ainsi que vous avez configuré votre interface python-pip . Si vous utilisez conda ou des environnements virtuels, suivez leurs recommandations pour l'installation des packages.
Cela devrait installer à la fois une interface de ligne de commande (CLI) et un package standard. Pour tester et voir si l'installation a réussi, vous pouvez essayer d'exécuter la commande :
sepal -h
Ce qui devrait imprimer le message d'aide associé à sépale. Si tout s'est bien passé pour vous jusqu'à présent, vous pouvez passer à la section exemple pour voir sepal en action !
L'utilisation recommandée de sepal se fait par l'interface de ligne de commande. Les simulations afin de calculer les temps de diffusion ainsi que l'analyse ou l'inspection ultérieure des résultats peuvent facilement être effectuées en tapant sepal suivi de run ou analyze . Le module analyze dispose de différentes options, pour visualiser les résultats ( inspect ), trier les profils en familles de motifs ( family ) ou encore soumettre les familles identifiées à une analyse d'enrichissement fonctionnel ( fea ). Pour une liste complète des commandes disponibles, faites sepal module -h , où module est l'un des run et analyze . Ci-dessous, nous illustrons comment le sépale peut être utilisé pour trouver des profils de transcription avec des modèles spatiaux.
Nous allons créer un dossier pour contenir nos résultats, qui fera également office de répertoire de travail. Depuis le répertoire principal du dépôt, faites :
cd res
mkdir example
cd exampleL’échantillon MOB sera utilisé pour illustrer notre analyse. Nous commençons par calculer les temps de diffusion pour chaque profil de transcription :
sepal run -c ../../data/real/mob.tsv.gz -mo 10 -mc 5 -o . -ar 1Vous trouverez ci-dessous un exemple (avec un affichage supplémentaire de la commande d'aide) de ce à quoi cela pourrait ressembler

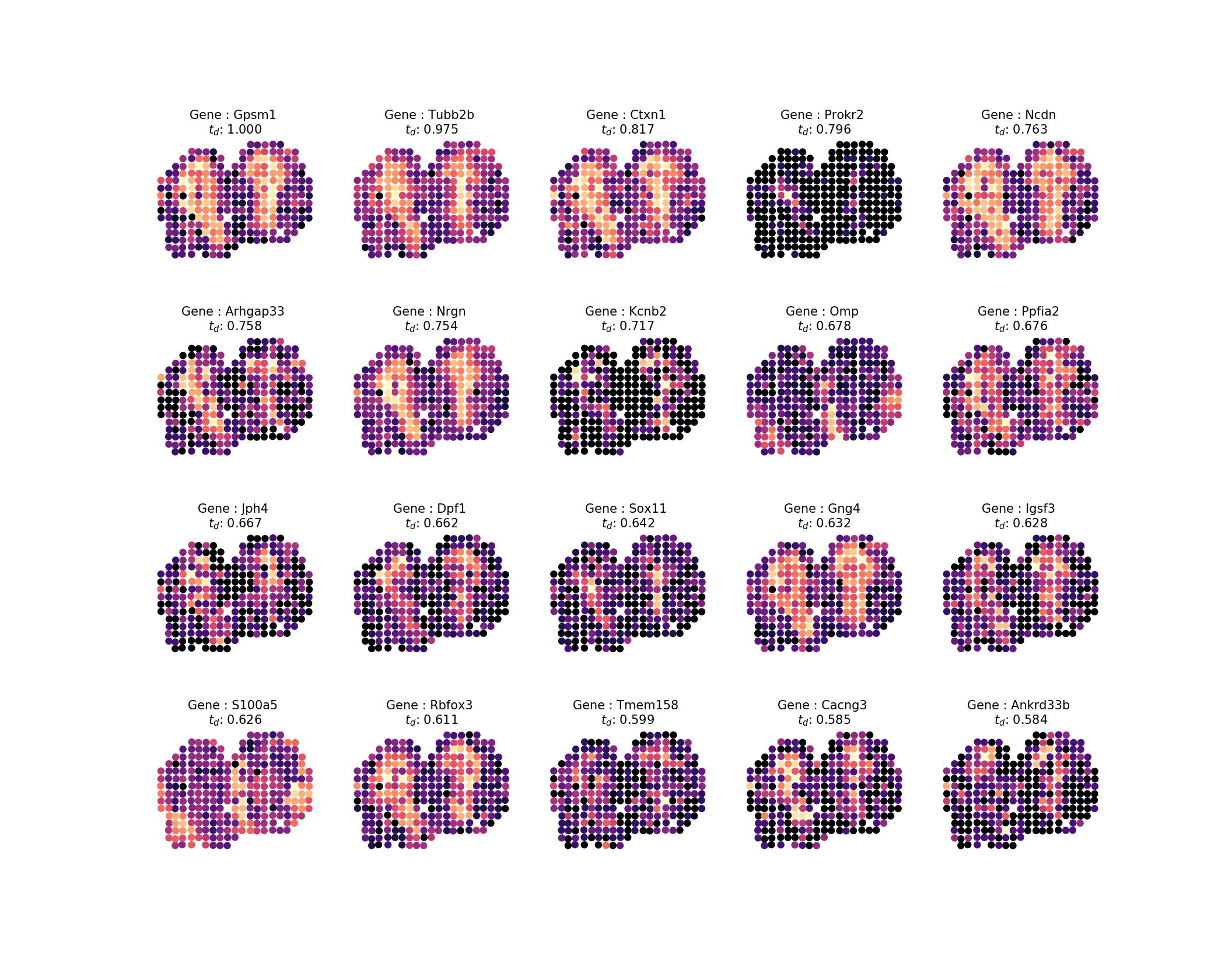
Après avoir calculé les temps de diffusion, nous voulons inspecter le résultat, comme dans l'étude, nous examinerons les 20 meilleurs profils. Nous pouvons facilement générer des images à partir de notre résultat en exécutant la commande :
sepal analyze -c ../../data/real/mob.tsv.gz
-r 20200409173043610345-top-diffusion-times.tsv
-ar 1k -o . inspect -ng 20 -nc 5Ce qui ressemblerait à ceci :

Le résultat sera l'image suivante :
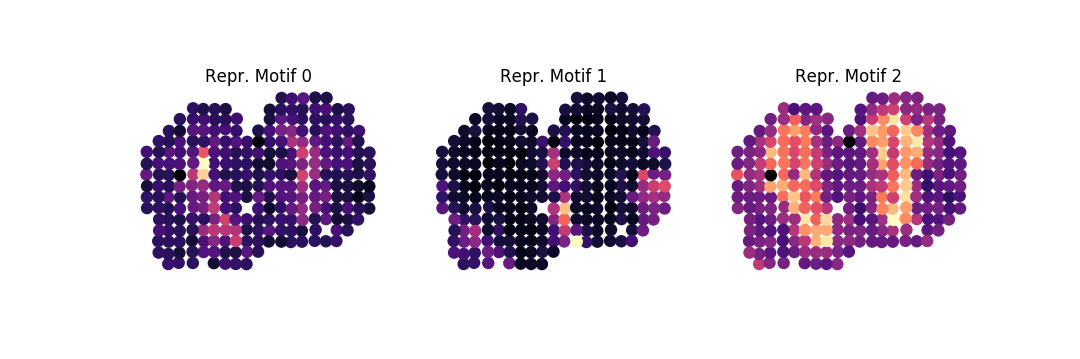
Ensuite, pour trier les 100 gènes les mieux classés dans un ensemble de familles de modèles, où 85 % de la variance de nos modèles devraient être expliqués par les modèles propres, procédez :
sepal analyze -c ../../data/real/mob.tsv.gz
-r 20200409173043610345-top-diffusion-times.tsv
-ar 1k -o . family -ng 100 -nbg 100 -eps 0.85 --plot -nc 3De là, nous obtenons les trois motifs représentatifs suivants pour chaque famille :
Nous pouvons soumettre nos familles à une analyse d'enrichissement, en exécutant :
sepal analyze -c ../../data/real/mob.tsv.gz
-r 20200409173043610345-top-diffusion-times.tsv
-ar 1k -o . fea -fl mob.tsv-family-index.tsv -or " mmusculus "où l'on voit par exemple que la Famille 2 est enrichie pour plusieurs processus liés à la fonction neuronale, à la génération et à la régulation :
| famille | indigène | nom | valeur_p | source | taille_intersection | |
|---|---|---|---|---|---|---|
| 2 | 2 | ALLER : 0007399 | développement du système nerveux | 0,00035977 | ALLER : BP | 26 |
| 3 | 2 | ALLER:0050773 | régulation du développement des dendrites | 0.000835883 | ALLER : BP | 8 |
| 4 | 2 | ALLER:0048167 | régulation de la plasticité synaptique | 0,00196494 | ALLER : BP | 8 |
| 5 | 2 | ALLER:0016358 | développement des dendrites | 0,00217167 | ALLER : BP | 9 |
| 6 | 2 | ALLER:0048813 | morphogenèse des dendrites | 0,00741589 | ALLER : BP | 7 |
| 7 | 2 | ALLER:0048814 | régulation de la morphogenèse des dendrites | 0,00800399 | ALLER : BP | 6 |
| 8 | 2 | ALLER:0048666 | développement des neurones | 0,0114088 | ALLER : BP | 16 |
| 9 | 2 | ALLER:0099004 | voie de signalisation de la kinase dépendante de la calmoduline | 0,0159572 | ALLER : BP | 3 |
| 10 | 2 | ALLER:0050804 | modulation de la transmission synaptique chimique | 0,0341913 | ALLER : BP | 10 |
| 11 | 2 | ALLER : 0099177 | régulation de la signalisation trans-synaptique | 0,0347783 | ALLER : BP | 10 |
Bien entendu, cette analyse n’est en aucun cas exhaustive. Mais plutôt un exemple rapide pour montrer comment on exploite la CLI pour sepal .
Bien que sepal ait été conçu comme un outil autonome, nous l'avons également construit pour qu'il soit fonctionnel en tant que package Python standard à partir duquel les fonctions peuvent être importées et utilisées dans un flux de travail intégré. Pour montrer comment cela peut être réalisé, nous fournissons un exemple reproduisant l’analyse du mélanome. D'autres exemples pourront être ajoutés ultérieurement.
L'entrée dans sepal doit être au format n_locations x n_genes , cependant si vos données sont structurées de la manière opposée ( n_genes x n_locations ), fournissez simplement l'indicateur --transpose lors de l'exécution de la simulation ou de l'analyse et cela sera pris en compte. de.
Nous prenons actuellement en charge les formats .csv , .tsv et .h5ad . Pour ce dernier, votre fichier doit être structuré selon CE format. Nous nous attendons à ce qu'il y ait une version de l'équipe scanpy dans un avenir proche, dans laquelle un format standardisé pour les données spatiales est présenté, mais d'ici là, nous utiliserons la norme susmentionnée.
Toutes les données réelles que nous avons utilisées sont publiques et sont accessibles via les liens suivants :
Les données synthétiques ont été générées par :
synthetic/img2cnt.pysynthetic/turing.pysynthetic/ablation.py Tous les résultats présentés dans l'étude se trouvent dans le dossier res , tant pour les données réelles que synthétiques. Pour chaque échantillon, nous avons structuré les résultats en conséquence :
res/sample-name/X-diffusion-times.tsv : temps de diffusion pour tous les gènes classésanalysis/ : contient la sortie de l'analyse secondaire

