reference_database_creator
bug fix --in-silico-pcr --untrimmed
CRABES ( C en train de manger R. bases de données de référence pour UN mplicon- B asséché S equencing) est un logiciel polyvalent qui génère des bases de données de référence organisées pour l’analyse métagénomique. Le flux de travail CRABS se compose de sept modules : (i) télécharger des données à partir de référentiels en ligne ; (ii) importer les données téléchargées au format CRABS ; (iii) extraire les régions d'amplicons par analyse PCR in silico ; (iv) récupérer des amplicons sans régions de liaison aux amorces grâce à des alignements avec des codes-barres extraits in silico ; (v) organiser et sous-ensembler la base de données locale via plusieurs paramètres de filtrage ; (vi) exporter la base de données locale dans divers formats selon les exigences du classificateur taxonomique ; et (vi) des fonctions de post-traitement, c'est-à-dire des visualisations, pour explorer et fournir un aperçu récapitulatif de la base de données de référence locale. Ces sept modules sont répartis en dix-huit fonctions et sont décrits ci-dessous. De plus, un exemple de code est fourni pour chacune des dix-huit fonctions. Enfin, un didacticiel permettant de créer une base de données locale de référence sur les requins pour l'ensemble d'amorces MiFish-E est fourni à la fin de ce document README pour fournir un exemple de script à titre de référence.
Nous sommes ravis d'annoncer que CRABS a connu une mise à jour majeure et une refonte du code basée sur les commentaires des utilisateurs, qui, nous l'espérons, amélioreront l'expérience utilisateur lors de la création de votre propre base de données de référence locale !
Veuillez trouver ci-dessous une liste des fonctionnalités et améliorations ajoutées à CRABS v 1.0.0 :
CRABS v 1.0.0 peut désormais être téléchargé manuellement en clonant ce référentiel GitHub (voir 4.1 Installation manuelle pour des informations détaillées). Nous mettrons à jour le conteneur Docker et le package conda dès que possible pour faciliter l'installation facile de la version la plus récente.
Lorsque vous utilisez CRABS dans vos projets de recherche, veuillez citer l'article suivant :
[Jeunen, G.-J., Dowle, E., Edgecombe, J., von Ammon, U., Gemmell, N. J., & Cross, H. (2022). crabs—A software program to generate curated reference databases for metabarcoding sequencing data. Molecular Ecology Resources, 00, 1– 14.](https://doi.org/10.1111/1755-0998.13741)
CRABS est une boîte à outils en ligne de commande uniquement fonctionnant sur des environnements Unix/Linux typiques et est exclusivement écrite en python3. Cependant, CRABS utilise le module de sous-processus en python pour exécuter plusieurs commandes en syntaxe bash afin de contourner les particularités spécifiques à Python et d'augmenter la vitesse d'exécution. Nous proposons trois façons d’installer CRABS. Pour la version la plus à jour de CRABS, nous recommandons l'installation manuelle en clonant ce référentiel GitHub et en installant 10 dépendances séparément (instructions d'installation pour toutes les dépendances fournies dans 4.1 Installation manuelle). CRABS peut également être installé via Docker et conda. Les deux méthodes permettent une installation facile en co-installant automatiquement toutes les dépendances. Notre objectif est de maintenir le conteneur Docker et le package conda à jour, même si un certain retard dans la mise à jour vers la version la plus récente peut survenir, en particulier pour le package conda. Vous trouverez ci-dessous les détails des trois approches.
Pour l'installation manuelle, clonez d'abord le référentiel CRABS. Cette étape nécessite que GitHub soit disponible sur la ligne de commande (instructions d'installation pour GitHub).
git clone https://github.com/gjeunen/reference_database_creator.git
En fonction de vos paramètres, CRABS devra peut-être être rendu exécutable sur votre système. Ceci peut être réalisé en utilisant le code ci-dessous.
chmod +x reference_database_creator/crabs
Une fois CRABS installé, nous devons nous assurer que toutes les dépendances sont installées et accessibles globalement. La dernière version de CRABS (version v 1.0.0 ) fonctionne sur Python 3.11.7 (ou toute version compatible avec 3.11.7) et s'appuie sur cinq modules Python qui pourraient ne pas être fournis en standard avec Python, ainsi que cinq logiciels externes. Toutes les dépendances sont répertoriées ci-dessous, ainsi qu'un lien vers les instructions d'installation. Les numéros de version fournis pour chaque module et logiciel sont ceux sur lesquels CRABS a été développé. Bien que des versions compatibles de chacun puissent également être utilisées.
Modules Python :
Logiciels externes :
Une fois CRABS et toutes les dépendances installées, CRABS peut être rendu accessible dans tout le système d'exploitation à l'aide du code ci-dessous.
export PATH="/path/to/crabs/folder:$PATH"
Remplacez /path/to/crabs/folder par le chemin réel vers le dossier du référentiel GitHub sur le système d'exploitation, c'est-à-dire le dossier créé lors de la commande git clone ci-dessus. L'ajout du code export au fichier .bash_profile ou .bashrc rendra CRABS accessible globalement à tout moment.
Docker est un projet open source qui permet le déploiement d'applications logicielles dans des « conteneurs » isolés de votre ordinateur et exécutés via un système d'exploitation hôte virtuel appelé Docker Engine. Le principal avantage de l’exécution de Docker sur les machines virtuelles est qu’elles utilisent beaucoup moins de ressources. Cette isolation signifie que vous pouvez exécuter un conteneur Docker sur la plupart des systèmes d'exploitation, notamment Mac, Windows et Linux. Vous devrez peut-être créer un compte gratuit pour utiliser Docker Desktop. Ce lien contient une belle introduction aux bases de l'utilisation de Docker. Voici un lien pour vous aider à démarrer et à vous orienter vers le multivers Docker.
Il n’y a que deux étapes pour faire fonctionner Crabs sur votre ordinateur. Tout d’abord, installez Docker Desktop sur votre ordinateur, qui est gratuit pour la plupart des utilisateurs. Voici les instructions pour Mac ; voici les instructions pour les ordinateurs Windows et voici les instructions pour Linux (la plupart des principales plates-formes Linux sont prises en charge). Une fois que Docker Desktop est installé et exécuté (l'application de bureau doit être en cours d'exécution pour que vous puissiez utiliser les commandes Docker sur la ligne de commande), il vous suffit de « extraire » notre image Crabs et vous êtes prêt à partir :
docker pull quay.io/swordfish/crabs:0.1.7
Bien que l'installation d'une application Docker soit simple, l'utilisation de ces applications peut être un peu délicate au début. Pour vous aider à démarrer, nous avons fourni quelques exemples de commandes utilisant la version docker de crabs. Ces exemples peuvent être trouvés dans le dossier docker_intro de ce dépôt . À partir de ces exemples, vous devriez être en mesure de configurer une base de données de référence complète et être prêt à partir. Nous continuerons à développer ces exemples et à tester cela dans de nombreuses situations différentes. Veuillez poser des questions et fournir des commentaires dans l'onglet Problèmes.
Pour installer le package conda, vous devez d’abord installer conda. Voir ce lien pour plus de détails. Si conda est déjà installé, il est recommandé de mettre à jour l'outil conda avec conda update conda avant d'installer CRABS.
Une fois conda installé, suivez les étapes ci-dessous pour installer CRABS et toutes les dépendances. Assurez-vous de saisir les commandes dans l’ordre dans lequel elles apparaissent ci-dessous.
conda create -n CRABS
conda activate CRABS
conda config --add channels bioconda
conda config --add channels conda-forge
conda install -c bioconda crabs
Une fois que vous avez entré la commande d'installation, conda traitera la demande (cela peut prendre environ une minute), puis affichera tous les packages et programmes qui seront installés et demandera de le confirmer. Tapez y pour démarrer l'installation. Une fois cette opération terminée, CRABS devrait être prêt à partir.
Nous avons testé cette installation sur les systèmes Mac et Linux. Nous n'avons pas encore testé sur le sous-système Windows pour Linux (WSL).
Utilisez le code ci-dessous pour vérifier si CRABS est installé avec succès et affichez les informations d'aide.
crabs -hLes informations d'aide répartissent les dix-huit fonctions en différents groupes, chaque groupe répertoriant la fonction en haut et les paramètres obligatoires et facultatifs en dessous.

CRABS contient sept modules, qui intègrent dix-huit fonctions :
Module 1 : télécharger des données à partir de référentiels en ligne
--download-taxonomy : télécharge les informations sur la taxonomie NCBI ;--download-bold : téléchargez les données de séquence à partir de la base de données Barcode of Life (BOLD) ;--download-embl : télécharger les données de séquence depuis l'European Nucleotide Archive (ENA ; EMBL) ;--download-mitofish : télécharge les données de séquence depuis la base de données MitoFish ;--download-ncbi : téléchargez les données de séquence depuis le National Center for Biotechnology Information (NCBI).Module 2 : importer les données téléchargées au format CRABS
--import : importez des séquences téléchargées ou des codes-barres personnalisés au format CRABS ;--merge : fusionne différents fichiers au format CRABS en un seul fichier.Module 3 : extraire les régions d'amplicons par analyse PCR in silico
--in-silico-pcr : extrait les amplicons des données téléchargées en localisant et en supprimant les régions de liaison aux amorces.Module 4 : récupérer des amplicons sans régions de liaison aux amorces
--pairwise-global-alignment : récupérez les amplicons sans régions de liaison aux amorces en alignant les séquences téléchargées sur les codes-barres extraits in silico .Module 5 : organiser et sous-ensembler la base de données locale via plusieurs paramètres de filtrage
--dereplicate : supprime les séquences en double ;--filter : supprime les séquences via plusieurs paramètres de filtrage ;--subset : sous-ensemble de la base de données locale pour conserver ou exclure les groupes taxonomiques spécifiés.Module 6 : exporter la base de données locale
--export : exporte la base de données au format CRABS vers différents formats selon les exigences du classificateur taxonomique à utiliser.Module 7 : fonctions de post-traitement pour explorer et fournir un aperçu synthétique de la base de données de référence locale
--diversity-figure : crée un graphique à barres horizontales affichant le nombre d'espèces et de groupes de séquences par niveau spécifié inclus dans la base de données de référence ;--amplicon-length-figure : crée un graphique linéaire illustrant les distributions de longueurs d'amplicons séparées par groupe taxonomique ;--phylogenetic-tree : crée un arbre phylogénétique avec des codes-barres issus de la base de données de référence pour une liste cible d'espèces ;--amplification-efficiency-figure : crée un graphique à barres affichant les mésappariements dans les régions de liaison aux amorces ;--completeness-table : crée une feuille de calcul contenant la disponibilité des codes-barres pour les groupes taxonomiques.Les données de séquençage initiales peuvent être téléchargées par CRABS à partir de quatre référentiels en ligne, dont (i) BOLD, (ii) EMBL, (iii) MitoFish et NCBI. A partir de la version v 1.0.0 , le téléchargement des données de chaque référentiel est découpé en fonction propre. De plus, CRABS ne formate pas automatiquement les données après le téléchargement pour augmenter la flexibilité et permettre le débogage en cas d'échec du téléchargement des données.
Outre le téléchargement des données de séquence, CRABS est également capable de télécharger les informations taxonomiques NCBI, que CRABS utilise pour créer la lignée taxonomique de chaque séquence.
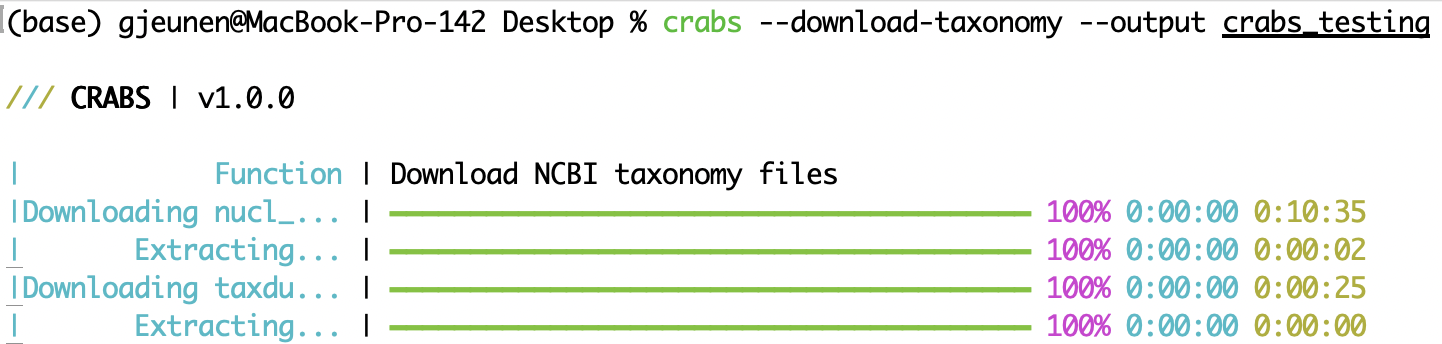
--download-taxonomy Pour attribuer une lignée taxonomique à chaque séquence téléchargée dans la base de données de référence (voir 5.2 Module 2), les informations taxonomiques doivent être téléchargées. CRABS utilise la taxonomie du NCBI et télécharge trois fichiers spécifiques sur votre ordinateur : (i) un fichier reliant les numéros d'accession aux identifiants taxonomiques ( nucl_gb.accession2taxid ), (ii) un fichier contenant des informations sur le nom phylogénétique associé à chaque identifiant taxonomique ( names.dmp ), et (iii) un fichier contenant des informations sur la manière dont les identifiants taxonomiques sont liés ( nodes.dmp ). Le répertoire de sortie des fichiers téléchargés peut être spécifié à l'aide du paramètre --output . Pour exclure soit le fichier nucl_gb.accession2taxid , soit les fichiers names.dmp et nodes.dmp , le paramètre --exclude acc2tax ou --exclude taxdump peut être fourni, respectivement. Le premier code ci-dessous ne télécharge aucun fichier, car acc2tax et taxdump sont fournis pour le paramètre --exclude . La deuxième ligne de code télécharge les trois fichiers dans le sous-répertoire --output crabs_testing . La capture d'écran ci-dessous affiche ce qui est imprimé sur la console lors de l'exécution de cette ligne de code.
crabs --download-taxonomy --exclude 'acc2taxid,taxdump'
crabs --download-taxonomy --output crabs_testing
--download-bold Les séquences BOLD sont téléchargées via le site Web BOLD. Le fichier de sortie, qui est structuré comme un document fasta de deux lignes, peut être spécifié à l'aide du paramètre --output . Les utilisateurs peuvent spécifier quel groupe taxonomique télécharger à l'aide du paramètre --taxon . Nous recommandons d'écrire une boucle for simple (exemple fourni ci-dessous) lorsque les utilisateurs souhaitent télécharger plusieurs groupes taxonomiques, limitant ainsi la quantité de données à télécharger depuis BOLD par instance. Cependant, si seul un nombre limité de groupes taxonomiques présente un intérêt, les noms de groupes taxonomiques peuvent également être séparés par | (exemple fourni ci-dessous). Nous recommandons également aux utilisateurs de vérifier si le nom du groupe taxonomique à télécharger est répertorié dans l'archive BOLD ou si des noms alternatifs doivent être utilisés. Par exemple, spécifier --taxon Chondrichthyes ne téléchargera pas toutes les séquences de poissons cartilagineux depuis BOLD, puisque ce nom de classe n'est pas répertorié sur BOLD. Les utilisateurs devraient plutôt utiliser --taxon Elasmobranchii dans ce cas. Les utilisateurs peuvent également spécifier de limiter le téléchargement à un marqueur génétique spécifique en fournissant le paramètre --marker . Lorsque plusieurs marqueurs génétiques présentent un intérêt, les noms des marqueurs doivent être séparés par | . Les quatre principaux marqueurs de codes-barres ADN sur BOLD sont COI-5P , ITS , matK et rbcL . La saisie du paramètre --marker est sensible à la casse.
Approche recommandée : une simple boucle for pour télécharger des données depuis BOLD pour plusieurs groupes taxonomiques (approche recommandée). Le code ci-dessous télécharge d'abord les données pour les Elasmobranchii, suivies des séquences attribuées aux Mammalia. Les données téléchargées seront écrites dans le sous-répertoire --output crabs_testing et placées dans deux fichiers distincts, indiquant quelles données appartiennent à quel groupe taxonomique, c'est-à-dire crabs_testing/bold_Elasmobranchii.fasta et crabs_testing/bold_Mammalia.fasta .
for taxon in Elasmobranchii Mammalia; do crabs --download-bold --taxon ${taxon} --output crabs_testing/bold_${taxon}.fasta; done

Option alternative : outre la boucle for recommandée, plusieurs noms de taxons peuvent être fournis simultanément en séparant les noms à l'aide de | .
crabs --download-bold --taxon 'Elasmobranchii|Mammalia' --output crabs_testing/bold_elasmobranchii_mammalia.fasta
--download-embl Les séquences de l'EMBL sont téléchargées via le site FTP de l'ENA. Les fichiers EMBL seront d'abord téléchargés au format « .fasta.gz » et seront automatiquement décompressés une fois le téléchargement terminé. Cette base de données n'offre pas autant de flexibilité en matière de téléchargement sélectif que BOLD ou NCBI. Les données de l'EMBL sont plutôt structurées en 15 divisions fiscales, qui peuvent être téléchargées séparément. La division fiscale à télécharger peut être spécifiée à l'aide du paramètre --taxon . Chaque division fiscale étant découpée en plusieurs fichiers, un * est prévu après le nom pour télécharger tous les fichiers. Les utilisateurs peuvent également télécharger un fichier spécifique en écrivant le nom du fichier au complet. Une liste des 15 options de division fiscale est fournie ci-dessous. Le répertoire de sortie et le nom du fichier peuvent être spécifiés à l'aide du paramètre --output .
Liste des divisions fiscales :
crabs --download-embl --taxon 'mam*' --output crabs_testing/embl_mam.fasta
--download-mitofish CRABS peut également télécharger la base de données MitoFish. Cette base de données est un seul fichier fasta de deux lignes. Le répertoire de sortie et le nom du fichier peuvent être spécifiés à l'aide du paramètre --output .
crabs --download-mitofish --output crabs_testing/mitofish.fasta

--download-ncbi Les séquences de la base de données NCBI sont téléchargées via les utilitaires de programmation Entrez. NCBI permet le téléchargement de données à partir de diverses bases de données, que les utilisateurs peuvent spécifier avec le paramètre --database . Pour la plupart des utilisateurs, la base de données --database nucleotide sera la plus appropriée pour créer une base de données de référence locale.
Pour spécifier les données à télécharger depuis NCBI, les utilisateurs proposent une recherche via le paramètre --query . Créer de bonnes recherches NCBI peut être difficile. Un bon moyen de créer une requête de recherche consiste à utiliser la fenêtre de recherche de la page Web NCBI. À partir de ce lien, effectuez d’abord une recherche initiale et appuyez sur Entrée. Cela vous amènera à la page de résultats où vous pourrez affiner davantage votre recherche. Dans la capture d'écran ci-dessous, nous avons encore affiné la recherche en limitant la longueur des séquences entre 100 et 25 000 pb et en incorporant uniquement des séquences mitochondriales. Les utilisateurs peuvent copier-coller le texte dans la zone « Détails de la recherche » sur le site Web et le fournir entre guillemets dans le paramètre --query . Un autre avantage de l'utilisation de la fenêtre de recherche de la page Web NCBI est que la page Web affichera le nombre de séquences correspondant à votre requête de recherche, qui doit correspondre au nombre de séquences signalées par CRABS. Cette page Web fournit un autre court didacticiel sur l'utilisation de la fonction de recherche sur la page Web NCBI que notre équipe a écrite pour des informations supplémentaires.
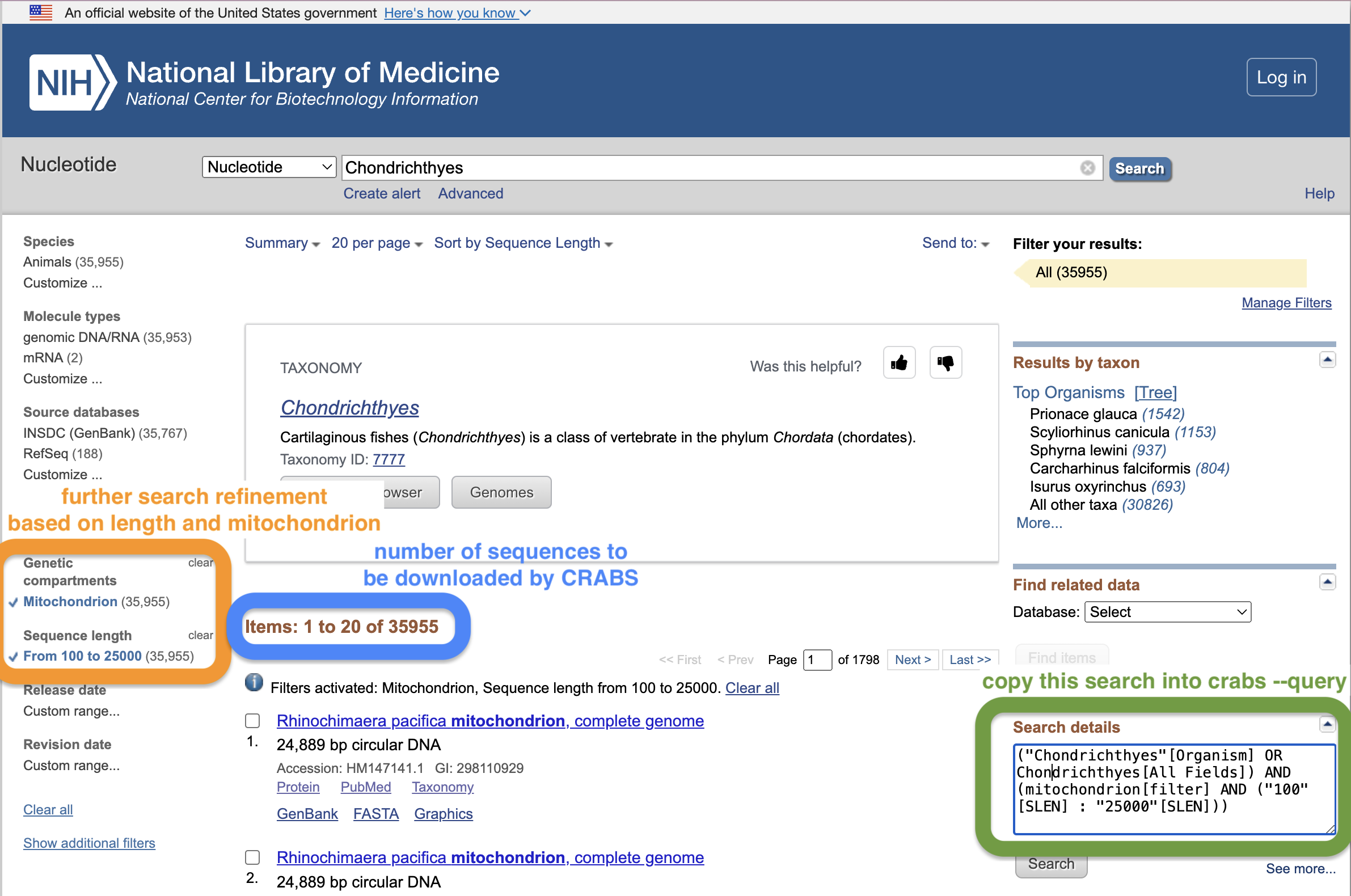
Outre la requête de recherche ( --query ), les utilisateurs peuvent restreindre davantage le terme de recherche en téléchargeant les données de séquence pour une liste d'espèces à l'aide du paramètre --species . Le paramètre --species prend soit une chaîne d'entrée de noms d'espèces séparés par + , soit un fichier .txt d'entrée avec un seul nom d'espèce par ligne dans le document. Le paramètre --batchsize offre aux utilisateurs la possibilité de télécharger des séquences par lots de N à partir du site Web du NCBI. Ce paramètre est par défaut de 5 000. Il n'est pas recommandé d'augmenter cette valeur au-dessus de 5 000, car les serveurs NCBI déconnecteront très probablement le téléchargement si trop de séquences sont téléchargées en même temps. Le paramètre --email permet aux utilisateurs de spécifier leur adresse e-mail, requise pour accéder aux serveurs NCBI. Enfin, le répertoire de sortie et le nom du fichier peuvent être spécifiés à l'aide du paramètre --output .
crabs --download-ncbi --query '("Chondrichthyes"[Organism] OR Chondrichthyes[All Fields]) AND (mitochondrion[filter] AND ("100"[SLEN] : "25000"[SLEN]))' --output crabs_testing/ncbi_chondrichthyes.fasta --email [email protected] --database nucleotide

--import Une fois les données des référentiels en ligne téléchargées, les fichiers devront être importés dans CRABS à l'aide de la fonction --import . Le format CRABS constitue une seule ligne délimitée par des tabulations par séquence contenant toutes les informations, y compris (i) l'ID de la séquence, (ii) le nom taxonomique analysé à partir du téléchargement initial, (iii) le numéro d'identification du taxon NCBI, (iv) la lignée taxonomique selon la taxonomie NCBI. , et (v) la séquence. CRABS tentera d'obtenir le numéro d'accession NCBI pour chaque séquence comme identifiant de séquence. Si la séquence ne contient pas de numéro d'accession, c'est-à-dire qu'elle n'est pas déposée sur NCBI, CRABS générera des identifiants de séquence uniques en utilisant le format suivant : crabs_*[num]*_taxonomic_name . Le format du document d'entrée est spécifié à l'aide du paramètre --import-format et spécifie le nom du référentiel à partir duquel les données ont été téléchargées, c'est-à-dire BOLD , EMBL , MITOFISH ou NCBI . La lignée taxonomique créée par CRABS est basée sur la taxonomie NCBI et CRABS nécessite les trois fichiers téléchargés à l'aide de la fonction --download-taxonomy , c'est-à-dire --names , --nodes et --acc2tax . Depuis la version v 1.0.0 , CRABS est capable de résoudre les synonymes et les noms non acceptés pour incorporer un plus grand nombre de séquences et de diversité dans la base de données de référence locale. Les rangs taxonomiques à inclure dans la lignée taxonomique peuvent être spécifiés à l'aide des paramètres --ranks . Bien que n'importe quel rang taxonomique puisse être inclus, nous vous recommandons d'utiliser l'entrée suivante pour inclure toutes les informations nécessaires pour la plupart des classificateurs taxonomiques --ranks 'superkingdom;phylum;class;order;family;genus;species' . Le fichier de sortie peut être spécifié à l'aide du paramètre --output et est un simple fichier .txt. Dans la fenêtre du terminal, CRABS imprime les résultats du nombre de séquences importées, ainsi que toutes les séquences pour lesquelles aucune lignée taxonomique n'a pu être générée.
crabs --import --import-format bold --input crabs_testing/bold_Elasmobranchii.fasta --names crabs_testing/names.dmp --nodes crabs_testing/nodes.dmp --acc2tax crabs_testing/nucl_gb.accession2taxid --output crabs_testing/crabs_bold.txt --ranks 'superkingdom;phylum;class;order;family;genus;species'

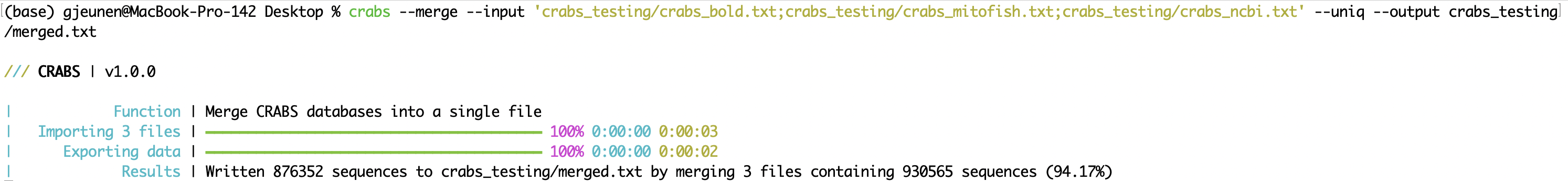
--merge Lorsque des données de séquence provenant de plusieurs référentiels en ligne sont téléchargées, les fichiers peuvent être fusionnés en un seul fichier après l'importation (voir 5.2.1 --import ) à l'aide de la fonction --merge . Les fichiers d'entrée à fusionner peuvent être saisis à l'aide du paramètre --input , les fichiers étant séparés par ; . Il est possible qu’une séquence ait été téléchargée plusieurs fois lors de son dépôt sur différents référentiels en ligne. L'utilisation du paramètre --uniq ne conserve qu'une seule version de chaque numéro d'accession. Le fichier de sortie peut être spécifié à l'aide du paramètre --output . Dans la fenêtre du terminal, CRABS imprime les résultats du nombre de séquences fusionnées, ainsi que le nombre de séquences conservées lors de l'utilisation du paramètre --uniq .
crabs --merge --input 'crabs_testing/crabs_bold.txt;crabs_testing/crabs_mitofish.txt;crabs_testing/crabs_ncbi.txt' --uniq --output crabs_testing/merged.txt
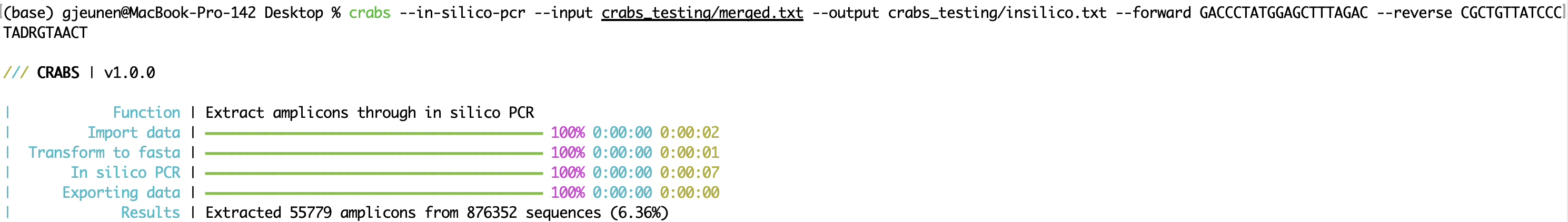
CRABS extrait la région amplicon de l'ensemble d'amorces en effectuant une PCR in silico (fonction : --in-silico-pcr ). CRABS utilise cutadapt v 4.4 pour la PCR in silico afin d'augmenter la vitesse d'exécution du code python traditionnel. Les noms des fichiers d'entrée et de sortie peuvent être spécifiés à l'aide des paramètres ' --input ' et ' --output ', respectivement. Les amorces directe et inverse doivent être fournies dans la direction 5'-3' en utilisant respectivement les paramètres ' --forward ' et ' --reverse '. CRABS inversera le complément de l'amorce inverse. Depuis la version v 1.0.0 , CRABS est capable de conserver les codes-barres dans les deux sens à l'aide d'une seule analyse PCR in silico . Par conséquent, aucune étape de complémentation inverse ni réexécution de la PCR in silico n’est effectuée, augmentant ainsi considérablement la vitesse d’exécution. Pour conserver les séquences pour lesquelles aucune région de liaison d'amorce n'a pu être trouvée, un fichier de sortie peut être spécifié pour le paramètre --untrimmed . Le nombre maximum autorisé de mésappariements trouvés dans les régions de liaison aux amorces peut être spécifié à l'aide du paramètre --mismatch , avec un paramètre par défaut de 4. Enfin, l'analyse PCR in silico peut être multithread dans CRABS. Par défaut, le nombre maximum de threads est utilisé, mais les utilisateurs peuvent spécifier le nombre de threads à utiliser avec le paramètre --threads .
crabs --in-silico-pcr --input crabs_testing/merged.txt --output crabs_testing/insilico.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT
Il est courant de supprimer les régions de liaison aux amorces des séquences de référence lorsqu’elles sont déposées dans une base de données en ligne. Par conséquent, lorsque la séquence de référence a été générée en utilisant la même amorce directe et/ou inverse que celle recherchée dans la fonction --in-silico-pcr , la fonction --in-silico-pcr n'aura pas réussi à récupérer la région de l'amplicon du séquence de référence. Pour tenir compte de cette possibilité, CRABS a la possibilité d'exécuter un alignement global par paire, mis en œuvre à l'aide de VSEARCH v 2.16.0 , pour extraire les régions d'amplicons pour lesquelles la séquence de référence ne contient pas l'intégralité des régions de liaison aux amorces directes et inverses. Pour ce faire, la fonction --pairwise-global-alignment récupère le fichier de base de données initialement téléchargé à l'aide du paramètre --input . La base de données dans laquelle effectuer la recherche est le fichier de sortie de --in-silico-pcr et peut être spécifiée à l'aide du paramètre --amplicons . Le fichier de sortie peut être spécifié à l'aide du paramètre --output . Les séquences d'amorces, utilisées uniquement pour calculer la longueur de la paire de bases, peuvent être définies avec les paramètres --forward et --reverse . Comme l'exécution de la fonction --pairwise-global-alignment peut prendre beaucoup de temps pour les bases de données volumineuses, la longueur de la séquence peut être limitée pour accélérer le processus à l'aide du paramètre --size-select . Le pourcentage minimum d'identité et de couverture des requêtes peut être spécifié à l'aide des paramètres --percent-identity et --coverage , respectivement. --percent-identity doit être fourni sous forme de pourcentage compris entre 0 et 1 (par exemple, 95 % = 0,95), tandis que --coverage doit être fourni sous forme de pourcentage compris entre 0 et 100 (par exemple, 95 % = 95). Par défaut, la fonction --pairwise-global-alignment est limitée à conserver les séquences où les séquences d'amorces ne sont pas entièrement présentes dans la séquence de référence (alignement commençant ou se terminant dans la longueur de l'amorce directe ou inverse). Lorsque le paramètre --all-start-positions est fourni, les résultats positifs seront inclus lorsque l'alignement est trouvé en dehors de la plage des régions de liaison d'amorce (manqué par la fonction --in-silico-pcr en raison d'un trop grand nombre de disparités dans le paramètre --all-start-positions). région de liaison à l'amorce). Nous ne recommandons pas d'utiliser --all-start-positions , car il est très peu probable qu'un code-barres soit amplifié à l'aide du jeu d'amorces spécifié de la fonction --in-silico-pcr lorsque plus de 4 non-concordances sont présentes dans l'amorce. régions de liaison.
crabs --pairwise-global-alignment --input crabs_testing/merged.txt --amplicons crabs_testing/insilico.txt --output crabs_testing/aligned.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT --size-select 10000 --percent-identity 0.95 --coverage 0.95
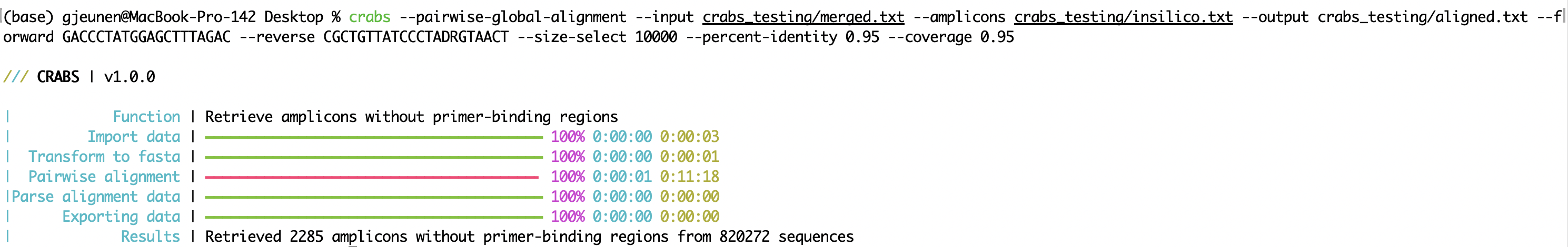
--pairwise-global-alignment L'exécution de la fonction --pairwise-global-alignment peut prendre beaucoup de temps lorsque CRABS traite des fichiers de séquence volumineux, même si le multithreading est pris en charge. Depuis la mise à jour vers CRABS v 1.0.0 , une structure de fichiers identique est en place de --import à --export , permettant ainsi aux fonctions d'être exécutées dans n'importe quel ordre. Bien que nous recommandons toujours de suivre l'ordre du flux de travail CRABS, la fonction --pairwise-global-alignment peut être considérablement accélérée lors de l'exécution des fonctions --dereplicate et --filter avant la fonction --in-silico-pcr . En exécutant ces étapes de curation avant --in-silico-pcr , le nombre de séquences devant être traitées par CRABS pour la fonction --pairwise-global-alignment sera considérablement réduit.
REMARQUE 1 : lors de l'exécution de la fonction --filter avant --in-silico-pcr , assurez-vous d'omettre tous les paramètres qui ont un impact direct sur la séquence, car --filter basera cela sur la séquence entière et non sur l'amplicon extrait. . Par conséquent, omettez les paramètres suivants : --minimum-length , --maximum-length , --maximum-n .
REMARQUE 2 : lors de l'exécution des fonctions --dereplicate et --filter avant --in-silico-pcr , il serait conseillé d'exécuter à nouveau les deux fonctions après --pairwise-global-alignment , car la base de données pourrait être davantage organisée maintenant que les amplicons sont extraits.
Une fois que tous les codes-barres potentiels pour l'ensemble d'amorces ont été extraits par les fonctions --in-silico-pcr et --pairwise-global-alignment , la base de données de référence locale peut subir une curation et un sous-ensemble supplémentaires dans CRABS à l'aide de diverses fonctions, notamment --dereplicate , --filter et --subset .
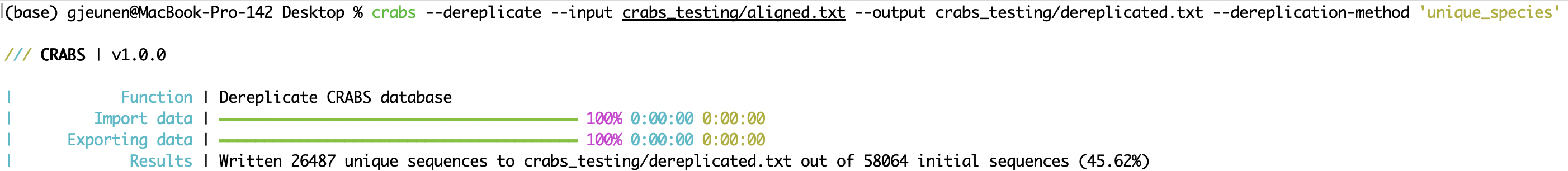
--dereplicate La première méthode de curation consiste à dérépliquer la base de données de référence locale à l'aide de la fonction --dereplicate . Il est possible que pour certains taxons, plusieurs codes-barres identiques soient contenus à ce stade dans la base de données de référence locale. Cela peut se produire lorsque différents groupes de recherche ont déposé des séquences identiques ou si la variation intra-spécifique entre les séquences d'un taxon n'est pas contenue dans le code-barres extrait. Il est préférable de supprimer ces codes-barres de référence identiques pour accélérer l'attribution de taxonomie, ainsi que pour améliorer les résultats d'attribution de taxonomie (en particulier pour les classificateurs taxonomiques fournissant un nombre limité de résultats, c'est-à-dire BLAST).
Les fichiers d'entrée et de sortie peuvent être spécifiés à l'aide des paramètres --input et --output , respectivement. CRABS propose trois méthodes de déréplication, qui peuvent être spécifiées à l'aide du paramètre --dereplication-method , notamment :
crabs --dereplicate --input crabs_testing/aligned.txt --output crabs_testing/dereplicated.txt --dereplication-method 'unique_species'
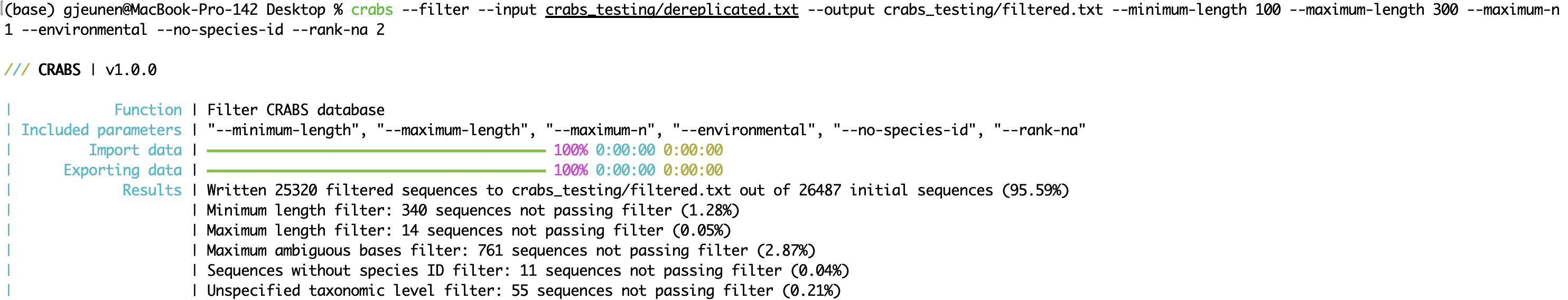
--filter La deuxième méthode de conservation consiste à filtrer la base de données de référence locale à l'aide de divers paramètres à l'aide de la fonction --filter . Les fichiers d'entrée et de sortie peuvent être spécifiés à l'aide des paramètres --input et --output , respectivement. À partir de la version v1.0.0 . CRABS intègre le filtrage basé sur six paramètres, dont :
--minimum-length : longueur de séquence minimale pour un amplicon à conserver dans la base de données ;--maximum-length : longueur de séquence maximale pour un amplicon à conserver dans la base de données ;--maximum-n : élimine les amplicons avec N bases ambiguës ou plus ( N );--environmental : supprime les séquences environnementales de la base de données ;--no-species-id : supprime les séquences pour lesquelles aucun nom d'espèce n'est disponible ;--rank-na : supprime les séquences avec N ou plus de niveaux taxonomiques non spécifiés. crabs --filter --input crabs_testing/dereplicated.txt --output crabs_testing/filtered.txt --minimum-length 100 --maximum-length 300 --maximum-n 1 --environmental --no-species-id --rank-na 2
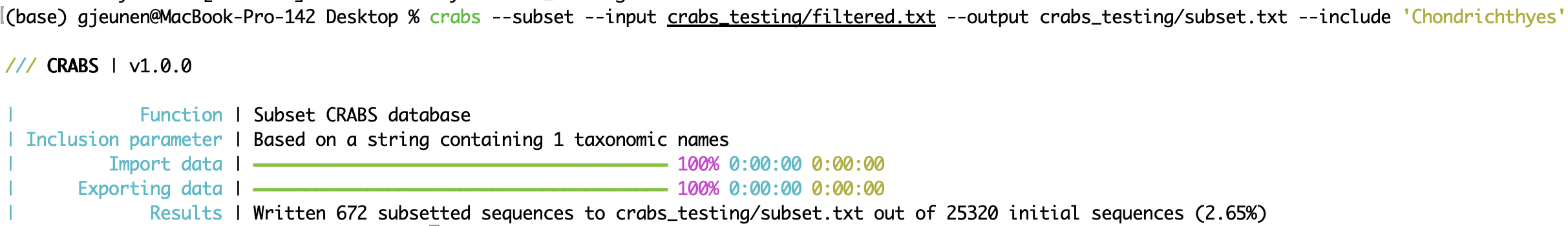
--subset La troisième et dernière méthode de conservation incorporée dans CRABS consiste à sous-ensembler la base de données de référence locale pour inclure (paramètre : --include ) ou exclure (paramètre : --exclude ) des taxons spécifiques à l'aide de la fonction --subset . Cette fonction permet de supprimer les codes-barres de référence des groupes taxonomiques sans intérêt pour la question de recherche. Ces groupes taxonomiques auraient pu être incorporés dans la base de données de référence locale en raison d'une amplification non spécifique potentielle de l'ensemble d'amorces. Un autre cas d'utilisation de --subset consiste à supprimer les séquences erronées connues.
Pour les classificateurs taxonomiques basés sur l'apprentissage automatique (IDTAXA) ou la distance k-mer (SINTAX), il peut être avantageux de sous-ensembler la base de données de référence en incluant uniquement les taxons connus pour être présents dans la région où les échantillons ont été prélevés et en excluant les espèces étroitement apparentées connues non connues. se produire dans la région pour augmenter la résolution taxonomique obtenue de ces classificateurs et obtenir de meilleurs résultats d'affectation de taxonomie.
Les fichiers d'entrée et de sortie peuvent être spécifiés à l'aide des paramètres --input et --output , respectivement. Les paramètres --include et --exclude peuvent contenir soit une liste de taxons séparés par ; ou un fichier .txt contenant un seul nom de taxon par ligne.
crabs --subset --input crabs_testing/filtered.txt --output crabs_testing/subset.txt --include 'Chondrichthyes'
Une fois la base de données de référence finalisée, elle peut être exportée dans différents formats pour répondre aux spécifications requises par la plupart des outils logiciels attribuant une taxonomie aux données métagénomiques. Les fichiers d'entrée et de sortie peuvent être spécifiés à l'aide des paramètres --input et --output , respectivement. A partir de la version v 1.0.0 , CRABS intègre le formatage de la base de données de référence pour six classificateurs différents (paramètre : --export-format ), dont :
--export-format 'sintax' : Le classificateur SINTAX est incorporé dans USEARCH et VSEARCH ;--export-format 'rdp' : Le classificateur RDP est un programme autonome largement utilisé dans les études sur le microbiome ;--export-format 'qiime-fasta' et --export-format 'qiime-text' : peuvent être utilisés pour attribuer un identifiant taxonomique dans QIIME et QIIME2 ;--export-format 'dada2-species' et --export-format 'dada2-taxonomy' : peuvent être utilisés pour attribuer un identifiant taxonomique dans DADA2 ;--export-format 'idt-fasta' et --export-format 'idt-text' : le classificateur IDTAXA est un algorithme d'apprentissage automatique incorporé dans le package DECIPHER R ;--export-format 'blast-notax' : crée une base de données de référence BLAST locale pour Blastn et Megablast où la sortie ne fournit pas d'identification taxonomique, mais répertorie le numéro d'accès;--export-format 'blast-tax' : Crée une base de données de référence BLAST locale pour Blastn et Megablast où la sortie fournit à la fois l'ID taxonomique et le numéro d'accès. crabs --export --input crabs_testing/subset.txt --output crabs_testing/BLAST_TAX_CHONDRICHTHYES --export-format 'blast-tax'

Lors de l'exportation de la base de données de référence locale vers un format unique (à l'exception des classificateurs où la base de données de référence est divisée sur plusieurs fichiers, c'est-à-dire Qiime, DADA2, idtaxa) suffira pour la plupart des utilisateurs, une simple boucle peut être écrite pour exporter le local Base de données de référence à plusieurs formats si les utilisateurs souhaitent comparer les résultats entre différents classificateurs taxonomiques. Un exemple est fourni ci-dessous pour exporter la base de données de référence locale dans les formats Sintax, RDP et IDTAXA.
for format in sintax.fasta rdp.fasta idt-fasta.fasta idt-text.txt; do crabs --export --input crabs_testing/subset.txt --output crabs_testing/chondrichthyes_${format} --export-format ${format%%.*}; done

Une fois la base de données de référence finalisée, les crabes peuvent exécuter cinq fonctions de post-traitement pour explorer et fournir un aperçu résumé de la base de données de référence locale, y compris (i) --diversity-figure ((II) - Figure de longueur (((((II) - Figure de longueur ((((II) - Figure de longueur (((((II) --amplicon-length-figure (((((II) iii) --phylogenetic-tree , (iv) --amplification-efficiency-figure et (v) --completeness-table .
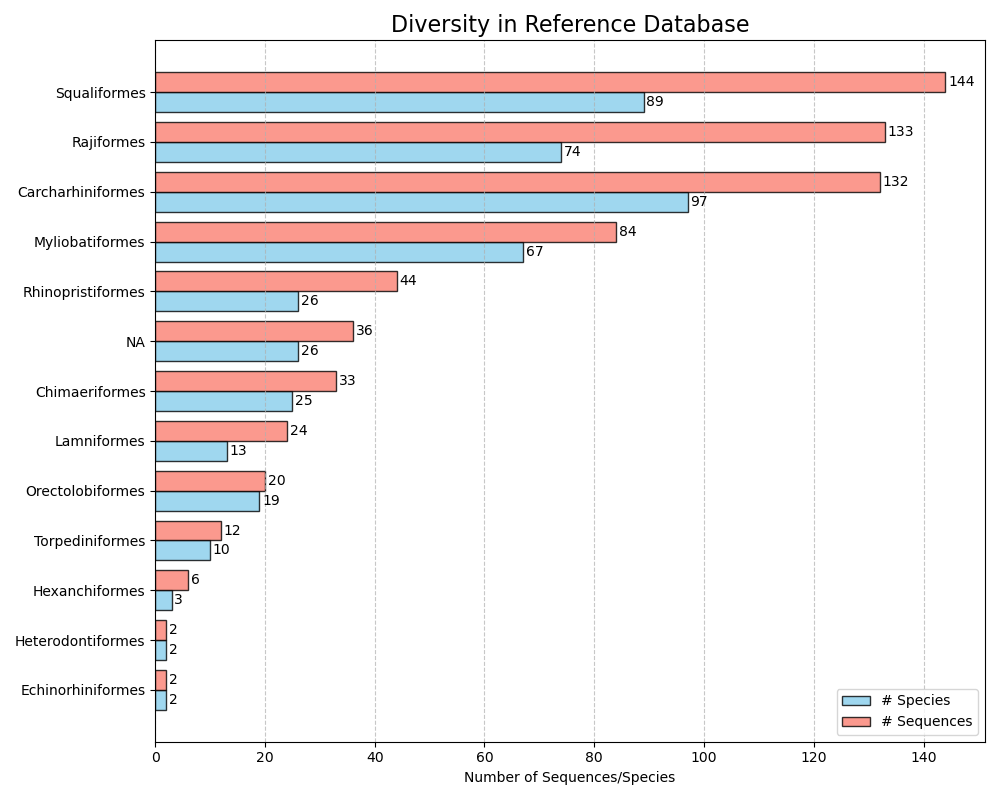
--diversity-figure La fonction --diversity-figure produit une parcelle de barre horizontale avec le nombre d'espèces (en bleu) et le nombre de séquences (en orange) par pour chaque groupe taxonomique dans la base de données de référence. L'utilisateur peut spécifier le rang taxonomique pour diviser la base de données de référence avec le paramètre --tax-level . Le niveau d'imposition est le nombre du rang dans lequel il est apparu lors de la fonction --import . Par exemple, si --ranks 'superkingdom;phylum;class;order;family;genus;species' a été utilisé pendant - la division --import en fonction de Superkingdom nécessiterait --tax-level 1 , phylum = --tax-level 2 , Class = --tax-level 3 , etc. Le fichier d'entrée au format Crabs peut être spécifié à l'aide du paramètre --input . La figure, au format .png, sera écrite dans le fichier de sortie, qui peut être spécifiée à l'aide du paramètre --output .
crabs --diversity-figure --input crabs_testing/subset.txt --output crabs_testing/diversity-figure.png --tax-level 4
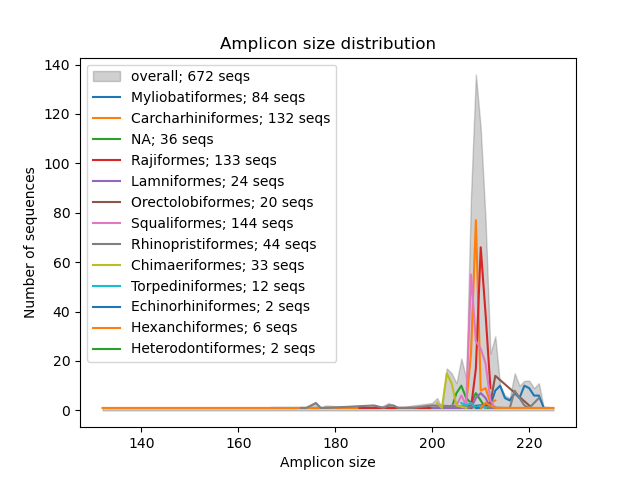
--amplicon-length-figure La fonction --amplicon-length-figure produit un graphique de ligne affichant la plage de la longueur d'amplicon. La plage globale de la longueur d'amplicon sur toutes les séquences de la base de données de référence est affichée dans une couleur gris ombragée, tandis que les résultats divisés par groupe taxonomique (paramètre: --tax-level ) sont superposés par des lignes colorées. De plus, la légende affiche le nombre de séquences attribuées à chacun des groupes taxonomiques et le nombre total de séquences dans la base de données de référence. Le fichier d'entrée au format CRABS peut être spécifié à l'aide du paramètre --input . La figure, au format .png, sera écrite dans le fichier de sortie, qui peut être spécifiée à l'aide du paramètre --output .
crabs --amplicon-length-figure --input crabs_testing/subset.txt --output crabs_testing/amplicon-length-figure.png --tax-level 4
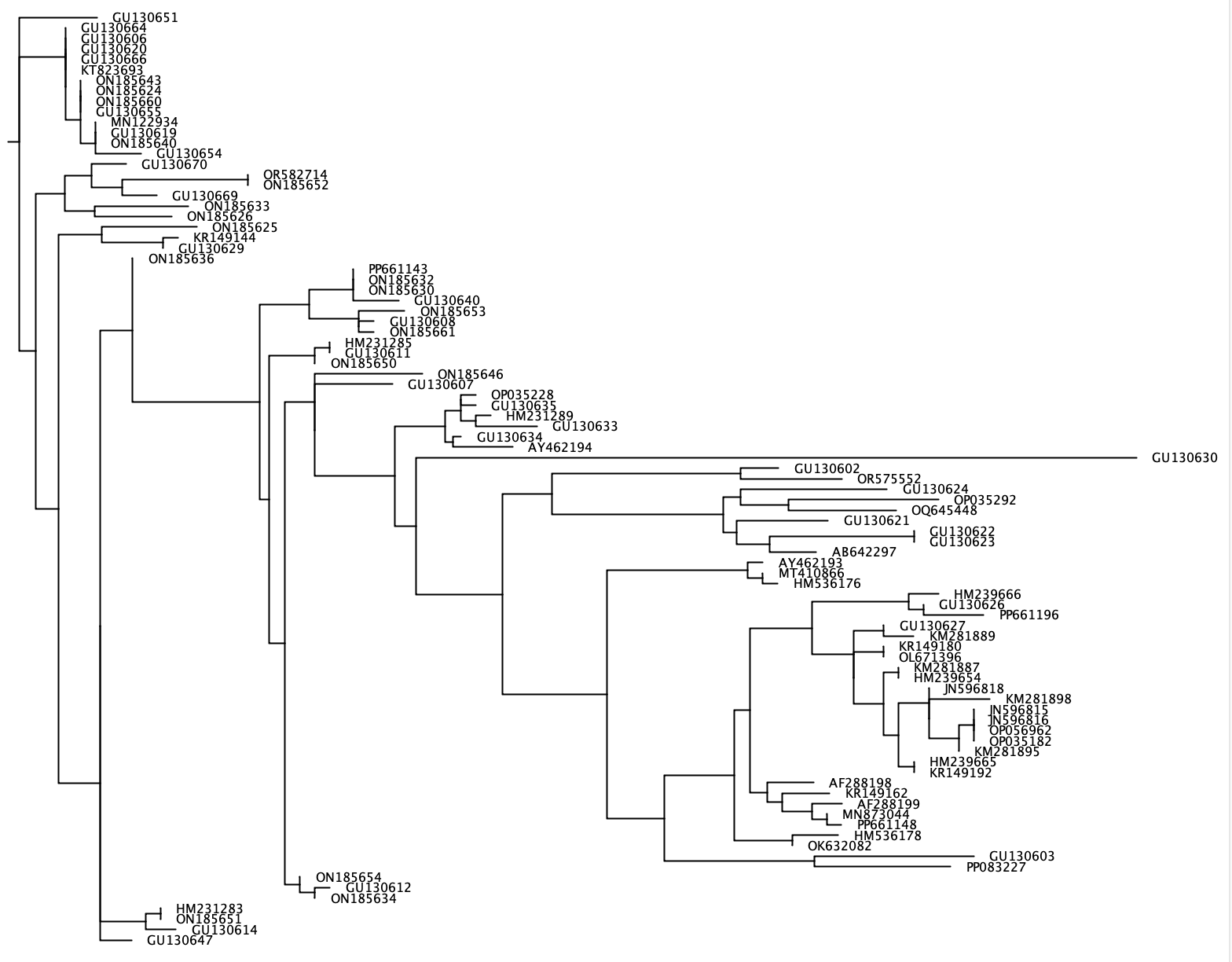
--phylogenetic-tree La fonction --phylogenetic-tree générera un arbre phylogénétique pour une liste d'espèces d'intérêt. Cette liste d'espèces d'intérêt peut être importée à l'aide du paramètre --species et se compose d'une chaîne d'entrée séparée par + ou un fichier .txt avec un seul nom d'espèce sur chaque ligne. Pour chaque espèce d'intérêt, les séquences seront extraites de la base de données de référence qui partagent un rang taxonomique défini par l'utilisateur (paramètre: --tax-level ) avec les espèces d'intérêt. Les crabes généreront un alignement de toutes les séquences extraites à l'aide de ClustalW2 V 2.1 et généreront un arbre phylogénétique de la jonction voisin à l'aide de FastTree. Cet arbre phylogénétique au format Newick sera écrit dans le fichier de sortie à l'aide du paramètre --output et peut être visualisé dans des logiciels tels que Figtree ou Geneious. Étant donné qu'un arbre phylogénétique séparé sera généré pour chaque espèce d'intérêt, --output prend un nom de fichier générique, tandis que le fichier de sortie exact contiendra ce nom générique suivi de '_Species_name.tree'.
crabs --phylogenetic-tree --input crabs_testing/subset.txt --output crabs_testing/phylo --tax-level 4 --species 'Carcharodon carcharias+Squalus acanthias'
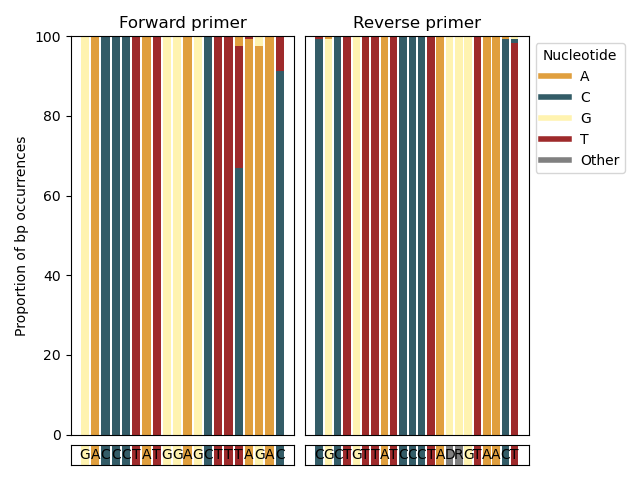
--amplification-efficiency-figure La fonction --amplification-efficiency-figure produira un graphique à barres, affichant la proportion de la paire de bases dans les régions de liaison aux amorces pour un groupe taxonomique spécifié par l'utilisateur, visualisant ainsi les lieux dans les régions de liaison à l'amorce avant et inverse où pourrait se produire dans le groupe taxonomique d'intérêt, influençant potentiellement l'efficacité de l'amplification. La fonction --amplification-efficiency-figure prend une base de données de référence Final Crabs Formatted comme entrée à l'aide du paramètre --amplicons . Pour trouver les informations sur les régions de liaison à l'amorce pour chaque séquence du fichier d'entrée, les séquences initialement téléchargées après l'importation doivent être fournies à l'aide du paramètre --input . Les séquences d'amorces avant et inverse (dans une direction 5 '- 3') sont fournies en utilisant les paramètres --forward et --reverse . Le nom du groupe taxonomique d'intérêt peut être fourni à l'aide du paramètre --tax-group et peut être défini à n'importe quel niveau taxonomique incorporé dans le fichier d'entrée. Enfin, le format figure dans .png sera écrit dans le fichier de sortie spécifié par le paramètre --output .
crabs --amplification-efficiency-figure --input crabs_testing/merged.txt --amplicons crabs_testing/subset.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT --output crabs_testing/amplification-efficiency.png --tax-group Carcharhiniformes
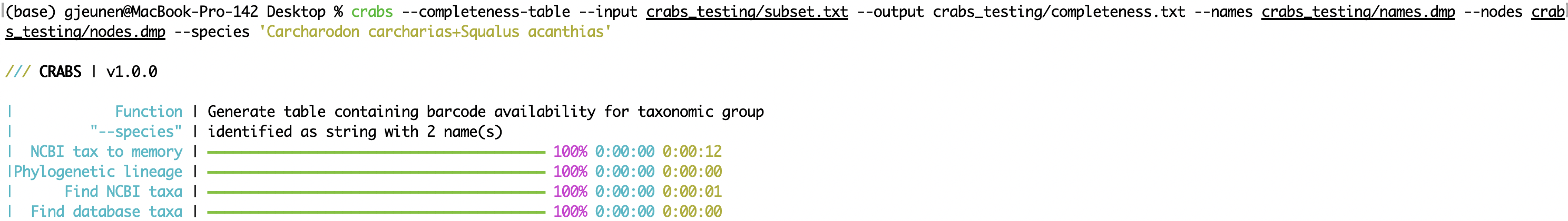
--completeness-table La fonction --completeness-table sortira un tableau délimité par l'onglet (paramètre: --output ) avec des informations sur une liste d'espèces d'intérêt. Cette liste d'espèces d'intérêt peut être importée à l'aide du paramètre --species et se compose d'une chaîne d'entrée séparée par + ou un fichier .txt avec un seul nom d'espèce sur chaque ligne. Une lignée taxonomique sera générée pour chaque espèce d'intérêt à l'aide des fichiers ' names.dmp ' et ' nœuds.dmp ' téléchargés à l'aide de la fonction --download-taxonomy à l'aide des paramètres --names et --nodes , respectivement. La table de sortie aura 10 colonnes fournissant les informations suivantes:
crabs --completeness-table --input crabs_testing/subset.txt --output crabs_testing/completeness.txt --names crabs_testing/names.dmp --nodes crabs_testing/nodes.dmp --species 'Carcharodon carcharias+Squalus acanthias'
crabs --version v 1.0.6 : correction de bogue -> analyse améliorée des en-têtes en gras pendant --import .crabs --version v 1.0.5 : correction de bogue -> Ajout d'une restriction de longueur à l'ID SEQ lors de la création de bases de données BLAST, au besoin pour le logiciel BLAST +.crabs --version v 1.0.4 : Informations ajoutées -> Informations correctes sur l'entrée de valeur pour --pairwise-global-alignment --coverage --percent-identity .crabs --version v 1.0.3 : correction de bogue -> Vérification de la réponse du serveur NCBI 3 fois avant de faire avorter l'analyse.crabs --version v 1.0.2 : correction de bogue -> capable de signaler lorsque 0 séquences est retournée après l'analyse.crabs --version v 1.0.1 : correction de bogue -> Courir la requête NCBI à succès à l'aide du paramètre --species .

