nucleotide transformer
1.0.0
Bienvenue sur ce référentiel InstaDeep Github, où sont présentés :
Nous sommes ravis de rendre ces travaux open source et de fournir à la communauté un accès au code et aux poids pré-entraînés pour ces neuf modèles de langage génomique et 2 modèles de segmentation. Les modèles du projet nucleotide transformer ont été développés en collaboration avec Nvidia et TUM, et les modèles ont été formés sur les nœuds DGX A100 de Cambridge-1. Le modèle du projet nucleotide transformer Agro a été développé en collaboration avec Google et le modèle formé sur les accélérateurs TPU-v4.
Dans l'ensemble, nos travaux fournissent de nouvelles informations liées au pré-entraînement et à l'application de modèles fondamentaux du langage, ainsi qu'à la formation de modèles les utilisant comme codeur de base, à la génomique avec de nombreuses possibilités d'applications dans ce domaine.
Dans ce référentiel, vous trouverez les éléments suivants :
Par rapport à d’autres approches, nos modèles intègrent non seulement les informations provenant de génomes de référence uniques, mais exploitent également les séquences d’ADN de plus de 3 200 génomes humains divers, ainsi que de 850 génomes provenant d’un large éventail d’espèces, y compris des organismes modèles et non modèles. Grâce à une évaluation robuste et approfondie, nous montrons que ces grands modèles fournissent une prédiction de phénotype moléculaire extrêmement précise par rapport aux méthodes existantes.
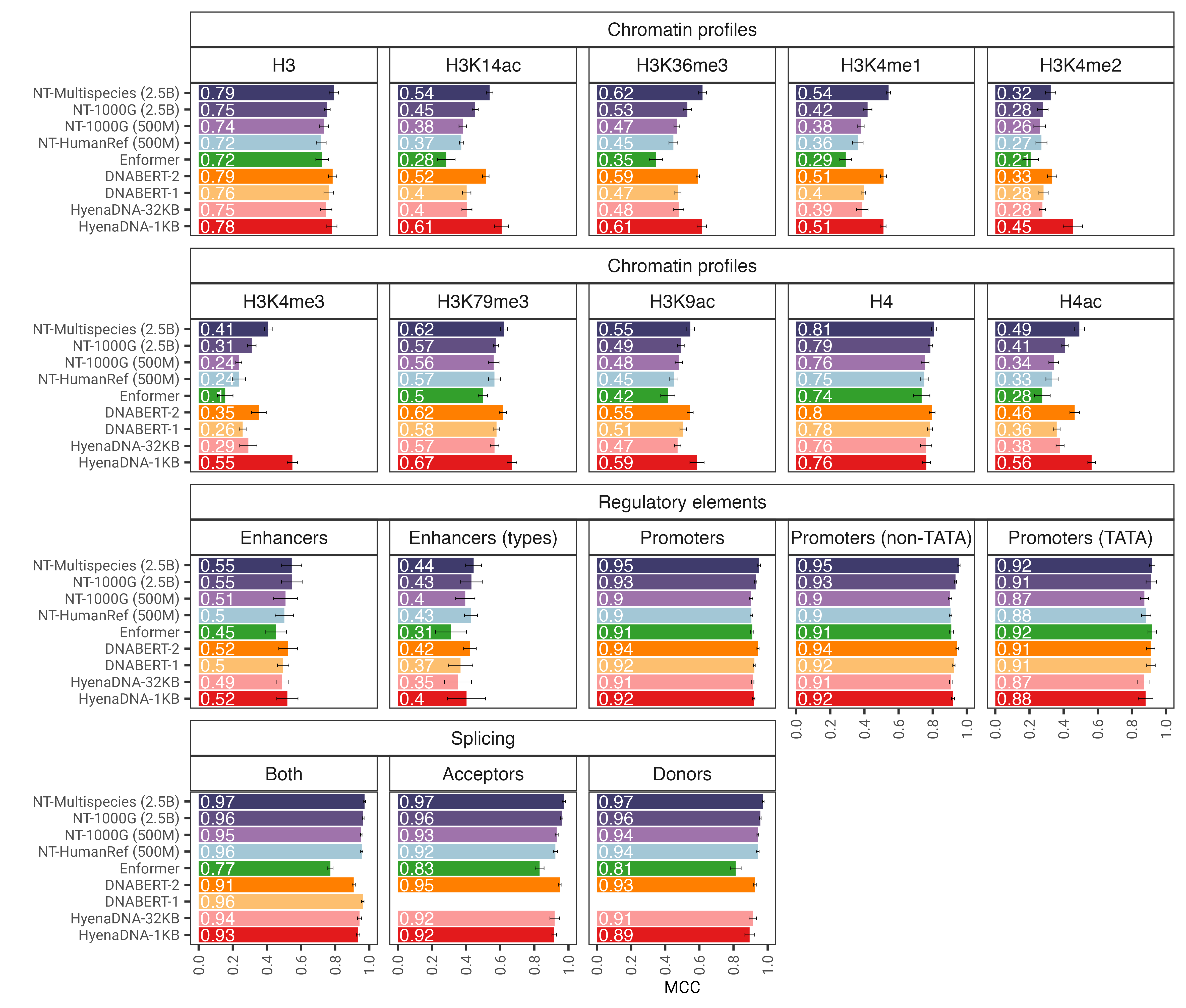
Fig. 1 : Le modèle nucleotide transformer prédit avec précision diverses tâches génomiques après un réglage fin. Nous montrons les résultats de performances dans les tâches en aval pour des modèles de transformateur affinés. Les barres d'erreur représentent 2 SD dérivées d'une validation croisée 10 fois.
Dans ce travail, nous présentons un nouveau modèle fondamental de grand langage formé sur les génomes de référence de 48 espèces végétales, avec un accent prédominant sur les espèces cultivées. Nous avons évalué les performances d'AgroNT sur plusieurs tâches de prédiction allant des caractéristiques de régulation au traitement de l'ARN et à l'expression des gènes, et avons montré qu'AgroNT peut obtenir des performances de pointe.
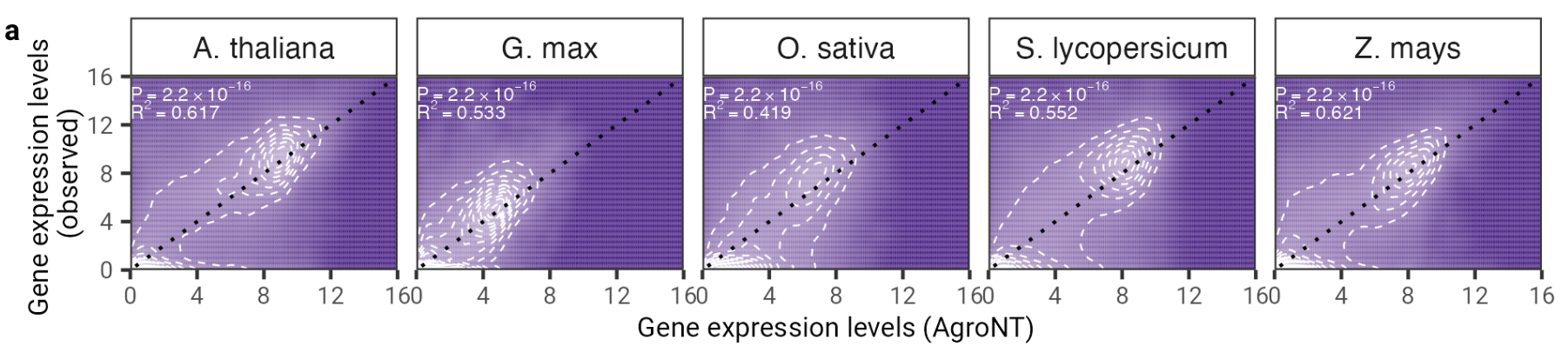
Fig. 2 : AgroNT fournit une prédiction de l'expression des gènes chez différentes espèces végétales. La prédiction de l’expression génique des gènes réfractaires dans tous les tissus est corrélée aux niveaux d’expression génique observés. Le coefficient de détermination (R 2 ) d'un modèle linéaire et les valeurs P associées entre les valeurs prédites et observées sont affichés.
Pour utiliser le code et les modèles pré-entraînés, il suffit :
pip install . .Vous pouvez ensuite télécharger et faire l'inférence avec l'un de nos neuf modèles en seulement quelques lignes de codes :
import haiku as hk
import jax
import jax . numpy as jnp
from nucleotide_transformer . pretrained import get_pretrained_model
# Get pretrained model
parameters , forward_fn , tokenizer , config = get_pretrained_model (
model_name = "500M_human_ref" ,
embeddings_layers_to_save = ( 20 ,),
max_positions = 32 ,
)
forward_fn = hk . transform ( forward_fn )
# Get data and tokenize it
sequences = [ "ATTCCGATTCCGATTCCG" , "ATTTCTCTCTCTCTCTGAGATCGATCGATCGAT" ]
tokens_ids = [ b [ 1 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens_str = [ b [ 0 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens = jnp . asarray ( tokens_ids , dtype = jnp . int32 )
# Initialize random key
random_key = jax . random . PRNGKey ( 0 )
# Infer
outs = forward_fn . apply ( parameters , random_key , tokens )
# Get embeddings at layer 20
print ( outs [ "embeddings_20" ]. shape )Les noms de modèles pris en charge sont :
Vous pouvez également exécuter nos modèles et trouver plus d'exemples de code dans Google Colab
Le code fonctionne à la fois sur GPU et TPU grâce à Jax !
Nos modèles nucleotide transformer v2 de deuxième version incluent une série de changements architecturaux qui se sont avérés plus efficaces : au lieu d'utiliser des intégrations positionnelles apprises, nous utilisons des intégrations rotatives qui sont utilisées à chaque couche d'attention et des unités linéaires fermées avec des activations brusques sans biais. Ces modèles améliorés acceptent également des séquences allant jusqu'à 2 048 jetons, ce qui conduit à une fenêtre contextuelle plus longue de 12 kbit/s. Inspirés par les lois d'échelle du Chinchilla, nous avons également entraîné nos modèles NT-v2 sur notre ensemble de données multi-espèces pour une durée plus longue (jetons 300B pour les modèles 50M et 100M ; jetons 1T pour les modèles 250M et 500M) par rapport aux modèles v1 (jetons 300B pour les quatre modèles).
Les couches de transformateur sont indexées 1, ce qui signifie que l'appel get_pretrained_model avec les arguments model_name="500M_human_ref" et embeddings_layers_to_save=(1, 20,) entraînera l'extraction des incorporations après la première et la 20ème couche de transformateur. Pour les transformateurs utilisant la tête Roberta LM, il est courant d'extraire les encastrements finaux après la norme de première couche de la tête LM plutôt qu'après le dernier bloc de transformateur. Par conséquent, si get_pretrained_model est appelé avec les arguments suivants embeddings_layers_to_save=(24,) , les intégrations ne seront pas extraites après la couche finale du transformateur mais plutôt après la norme de la première couche de la tête LM.
Les modèles SegmentNT exploitent un transformateur nucleotide transformer (NT) dont nous avons retiré la tête du modèle de langage et l'avons remplacée par une tête de segmentation U-Net unidimensionnelle pour prédire l'emplacement de plusieurs types d'éléments génomiques dans une séquence avec une résolution de nucléotide unique. Nous présentons deux variantes de modèles différentes sur 14 classes différentes d'éléments génomiques humains dans des séquences d'entrée allant jusqu'à 30 Ko. Ceux-ci incluent les gènes (gènes codant pour les protéines, lncARN, 5'UTR, 3'UTR, exon, intron, accepteurs d'épissage et sites donneurs) et régulateurs (signal polyA, promoteurs et amplificateurs invariants et spécifiques aux tissus, et liés au CTCF). sites) éléments. SegmentNT atteint des performances supérieures par rapport à l'architecture de segmentation U-Net de pointe, bénéficiant des poids pré-entraînés de NT, et démontre une généralisation sans tir jusqu'à 50 kbit/s.
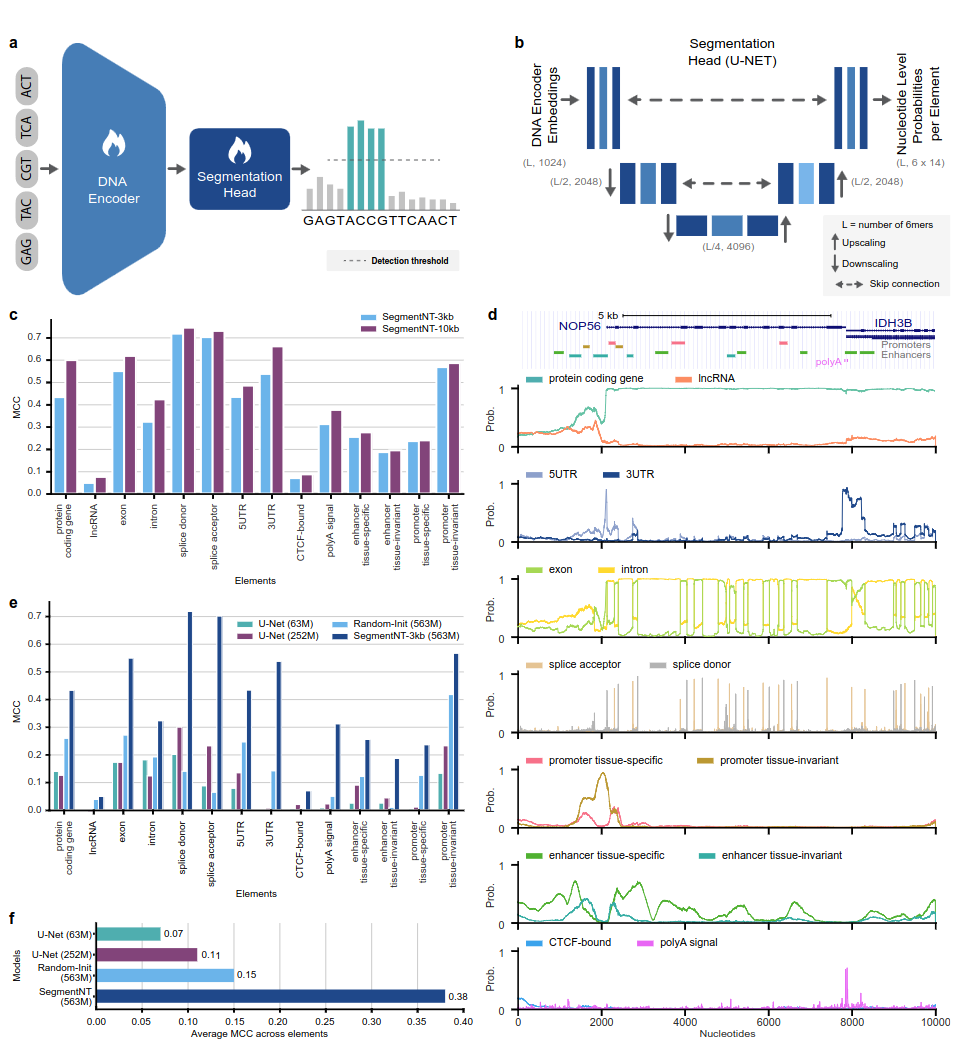
Fig. 1 : SegmentNT localise les éléments génomiques à une résolution nucléotidique.
Pour utiliser le code et les modèles pré-entraînés, il suffit :
pip install . .Vous pouvez ensuite télécharger et déduire une séquence avec n'importe lequel de nos modèles en seulement quelques lignes de codes :
rescaling factor est défini sur celui utilisé lors de la formation. Dans le cas où vous devez déduire sur des séquences comprises entre 30 kbp et 50 kbp, assurez-vous de passer l'argument rescaling_factor dans la fonction get_pretrained_segment_nt_model avec la valeur rescaling_factor = max_num_nucleotides / max_num_tokens_nt où num_dna_tokens_inference est le nombre de jetons à l'inférence (soit 6669 pour une séquence de 40008). paires de bases) et max_num_tokens_nt est le nombre maximum de jetons sur lesquels le transformateur de nucléotide du squelette a été formé, c'est-à-dire 2048 .
? Le cahier examples/inference_segment_nt.ipynb montre comment déduire une séquence de 50 Ko et tracer les probabilités pour reproduire la figure 3 de l'article.
? Les modèles SegmentNT ne gèrent aucun « N » dans la séquence d'entrée car chaque nucléotides doit être tokenisé en 6-mers, ce qui ne peut pas être le cas lors de l'utilisation de séquences contenant une ou plusieurs paires de bases « N ».
import haiku as hk
import jax
import jax . numpy as jnp
from nucleotide_transformer . pretrained import get_pretrained_segment_nt_model
# Initialize CPU as default JAX device. This makes the code robust to memory leakage on
# the devices.
jax . config . update ( "jax_platform_name" , "cpu" )
backend = "cpu"
devices = jax . devices ( backend )
num_devices = len ( devices )
print ( f"Devices found: { devices } " )
# The number of DNA tokens (excluding the CLS token prepended) needs to be dividible by
# 2 to the power of the number of downsampling block, i.e 4.
max_num_nucleotides = 8
assert max_num_nucleotides % 4 == 0 , (
"The number of DNA tokens (excluding the CLS token prepended) needs to be dividible by"
"2 to the power of the number of downsampling block, i.e 4." )
parameters , forward_fn , tokenizer , config = get_pretrained_segment_nt_model (
model_name = "segment_nt" ,
embeddings_layers_to_save = ( 29 ,),
attention_maps_to_save = (( 1 , 4 ), ( 7 , 10 )),
max_positions = max_num_nucleotides + 1 ,
)
forward_fn = hk . transform ( forward_fn )
apply_fn = jax . pmap ( forward_fn . apply , devices = devices , donate_argnums = ( 0 ,))
# Get data and tokenize it
sequences = [ "ATTCCGATTCCGATTCCAACGGATTATTCCGATTAACCGATTCCAATT" , "ATTTCTCTCTCTCTCTGAGATCGATGATTTCTCTCTCATCGAACTATG" ]
tokens_ids = [ b [ 1 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens_str = [ b [ 0 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens = jnp . asarray ( tokens_ids , dtype = jnp . int32 )
random_key = jax . random . PRNGKey ( seed = 0 )
keys = jax . device_put_replicated ( random_key , devices = devices )
parameters = jax . device_put_replicated ( parameters , devices = devices )
tokens = jax . device_put_replicated ( tokens , devices = devices )
# Infer on the sequence
outs = apply_fn ( parameters , keys , tokens )
# Obtain the logits over the genomic features
logits = outs [ "logits" ]
# Transform them in probabilities
probabilities = jnp . asarray ( jax . nn . softmax ( logits , axis = - 1 ))[..., - 1 ]
print ( f"Probabilities shape: { probabilities . shape } " )
print ( f"Features inferred: { config . features } " )
# Get probabilities associated with intron
idx_intron = config . features . index ( "intron" )
probabilities_intron = probabilities [..., idx_intron ]
print ( f"Intron probabilities shape: { probabilities_intron . shape } " )Les noms de modèles pris en charge sont :
Le code fonctionne à la fois sur GPU et TPU grâce à Jax !
Les modèles sont formés sur des séquences d'une longueur maximale de 1 000 jetons, y compris le jeton <CLS> ajouté automatiquement au début de la séquence. Le tokenizer commence la tokenisation de gauche à droite en regroupant les lettres « A », « C », « G » et « T » en 6-mers. La lettre "N" est choisie pour ne pas être regroupée à l'intérieur des k-mers, donc chaque fois que le tokenizer rencontre un "N", ou si le nombre de nucléotides dans la séquence n'est pas un multiple de 6, il tokenisera les nucléotides sans regroupement. eux. Des exemples sont donnés ci-dessous :
dna_sequence_1 = "ACGTGTACGTGCACGGACGACTAGTCAGCA"
tokenized_dna_sequence_1 = [ < CLS > , < ACGTGT > , < ACGTGC > , < ACGGAC > , < GACTAG > , < TCAGCA > ]
dna_sequence_2 = "ACGTGTACNTGCACGGANCGACTAGTCTGA"
tokenized_dna_sequence_2 = [ < CLS > , < ACGTGT > , < A > , < C > , < N > , < TGCACG > , < G > , < A > , < N > , < CGACTA > , < GTCTGA > ]Tous les transformateurs v1 et v2 peuvent donc prendre des séquences allant respectivement jusqu'à 5994 et 12282 nucléotides s'il n'y a pas de « N » à l'intérieur.
La collection de modèles présentés dans ce référentiel est disponible sur les espaces huggingface d'Instadeep ici : L'espace du nucleotide transformer et l'espace nucleotide transformer !
Nous remercions Maša Roller, ainsi que les membres du Rostlab, en particulier Tobias Olenyi, Ivan Koludarov et Burkhard Rost, pour les discussions constructives qui ont permis d'identifier des directions de recherche intéressantes. Nous exprimons également notre gratitude à tous ceux qui déposent des données expérimentales dans des bases de données publiques, à ceux qui maintiennent ces bases de données et à ceux qui mettent gratuitement à disposition des méthodes analytiques et prédictives. Nous remercions également l'équipe de développement de Jax.
Si vous trouvez ce référentiel utile dans votre travail, veuillez ajouter une citation pertinente à l'un de nos articles associés :
Le papier nucleotide transformer :
@article { dalla2023nucleotide ,
title = { The nucleotide transformer : Building and Evaluating Robust Foundation Models for Human Genomics } ,
author = { Dalla-Torre, Hugo and Gonzalez, Liam and Mendoza Revilla, Javier and Lopez Carranza, Nicolas and Henryk Grywaczewski, Adam and Oteri, Francesco and Dallago, Christian and Trop, Evan and Sirelkhatim, Hassan and Richard, Guillaume and others } ,
journal = { bioRxiv } ,
pages = { 2023--01 } ,
year = { 2023 } ,
publisher = { Cold Spring Harbor Laboratory }
}Papier nucleotide transformer agronucléotidique :
@article { mendoza2024foundational ,
title = { A foundational large language model for edible plant genomes } ,
author = { Mendoza-Revilla, Javier and Trop, Evan and Gonzalez, Liam and Roller, Ma{v{s}}a and Dalla-Torre, Hugo and de Almeida, Bernardo P and Richard, Guillaume and Caton, Jonathan and Lopez Carranza, Nicolas and Skwark, Marcin and others } ,
journal = { Communications Biology } ,
volume = { 7 } ,
number = { 1 } ,
pages = { 835 } ,
year = { 2024 } ,
publisher = { Nature Publishing Group UK London }
}Papier SegmentNT
@article { de2024segmentnt ,
title = { SegmentNT: annotating the genome at single-nucleotide resolution with DNA foundation models } ,
author = { de Almeida, Bernardo P and Dalla-Torre, Hugo and Richard, Guillaume and Blum, Christopher and Hexemer, Lorenz and Gelard, Maxence and Pandey, Priyanka and Laurent, Stefan and Laterre, Alexandre and Lang, Maren and others } ,
journal = { bioRxiv } ,
pages = { 2024--03 } ,
year = { 2024 } ,
publisher = { Cold Spring Harbor Laboratory }
}Si vous avez des questions ou des commentaires sur le code et les modèles, n'hésitez pas à nous contacter.
Merci de votre intérêt pour notre travail !


