poseidon
1.0.3

Veuillez noter que le code de Poséidon vient d'être transféré sur NextFlow afin qu'il puisse y avoir encore des bogues. N'hésitez pas à signaler les problèmes!
Ici, nous présentons Poséidon, un pipeline pour détecter des sites sélectionnés significativement positifs et des événements de recombinaison possibles dans un alignement de plusieurs séquences codant pour les protéines. Les sites qui subissent une sélection positive fournissent des informations dans l'histoire évolutive de vos séquences, par exemple, montrant des points chauds d'importants mutations, accumulés comme les résultats des races d'armes à hôte viral pendant l'évolution.
Poséidon s'appuie sur une variété d'outils tiers différents (voir ci-dessous). Mais ne vous inquiétez pas, nous avons encapsulé chaque outil dans son propre conteneur Docker et les avons connectés dans le système de gestion du flux de travail NextFlow.
Allez directement à un petit exemple de sortie de Poséidon pour la protéine de pointe SARS-COV-2 par rapport à une étude récente de Zhou et al . 2020.
Vous n'avez besoin que de NextFlow (version 20. +) et Docker installé pour exécuter le pipeline. Toutes les dépendances seront tirées automatiquement.
Soit exécuter Poseidon en clonant ce référentiel:
Git Clone https://github.com/hoelzer/poseidon.gitcd Poseidon NextFlow Run Poseidon.nf --Help
ou laissez Nextflow faire la traction
NextFlow Pull Hoelzer / Poseidon
Nous vous recommandons d'utiliser une version spécifique de Poseidon via
#PullNextflow Pull Hoelzer / Poseidon -r V1.0.1 # Runnextflow Run Hoelzer / Poseidon -r V1.0.1 - Help
Selon votre procédure d'installation, mettez à jour le pipeline via git pull ou nextflow pull hoelzer/poseidon .
Important: Poséidon a besoin de séquences nucléotidiques avec un cadre de lecture ouvert correct comme entrée. De plus, les résultats dépendent fortement de votre sélection de séquences, vous pouvez donc envisager d'exécuter le pipeline plusieurs fois avec différents échantillonnages de vos séquences d'entrée. De plus, le pipeline ne peut pas fonctionner avec trop de séquences car dans son cœur, Poseidon utilise le codeml de la suite PAML qui n'est pas entré pour> 100 séquences. Veuillez trouver une description détaillée des paramètres d'entrée et des paramètres ci-dessous.
NextFlow peut être facilement exécuté sur différents environnements comme votre machine locale, un cluster haute performance ou le cloud. Différents -profile sont utilisés pour indiquer à NextFlow quel système doit être utilisé. Pour l'exécution locale -profile local,docker doit être utilisé (et est également la valeur par défaut). Vous pouvez également exécuter Poséidon sur un HPC en utilisant la singularité via -profile lsf,singularity , -profile slurm,singularity ou -profile sge,singularity . Dans de tels cas, veuillez également envisager de s'ajuster --cachedir pour indiquer où stocker des images de singularité sur votre cluster. Le paramètre --workdir pourrait également être utile pour ajuster où stocker les répertoires de travail temporaires (par exemple, utiliser /scratch au lieu de /tmp en fonction de votre configuration HPC.)
Supposons maintenant que vous avez utilisé NextFlow pour extraire le code Poseidon et que vous exécutez le pipeline sur une machine locale en utilisant le profil par défaut -profile local,docker .
# Afficher l'aide nextflow Run Hoelzer / Poseidon - help # Exécutez un petit exemple sur une machine locale avec # (la première fois, cela aura besoin de plus de temps car les conteneurs Docker sont téléchargés) NextFlow Run Hoelzer / Poseidon -R V1.0.1 - Fasta ~ / .Nextflow / Assets / Hoelzer / Poseidon / test_data / bats_mx1_small.fasta --Cores 4 # reprendre un runnextflow run run hoelzer / poseidon -r v1.0.1 --fasta ~ / .nextflow / actifs / hoelzer / poseidon / test_data / bats_mx1_small.fasta --Cores 4-résume # Au lieu d'utiliser tous les cœurs disponibles, n'utilisez qu'une quantité maximale sur le machineneNExtflow Local Run Hoelzer / Poseidon -R V1.0.1 --FASTA ~ / .Nextflow / Assets / Hoelza / Poseidon / Test_Data / BATS_MX1_SMALL.Fasta --max_cores 8 - Cores 4
Pour reproduire les résultats de sélection positifs rapportés dans Fuchs et al . (2017), Journal of Virology Run:
NextFlow Run Hoelzer / Poseidon -R V1.0.0 --FASTA ~ / .Nextflow / Assets / Hoelzer / Poseidon / Test_Data / Bats_MX1.Fasta --Cores 4 --kh --outgroup "Pteropus_Alecto, Eidolon_helvum, Rouseettus_aegyptiacus, hysignatus_monstrosus" --reference "myotis_daubentonii"
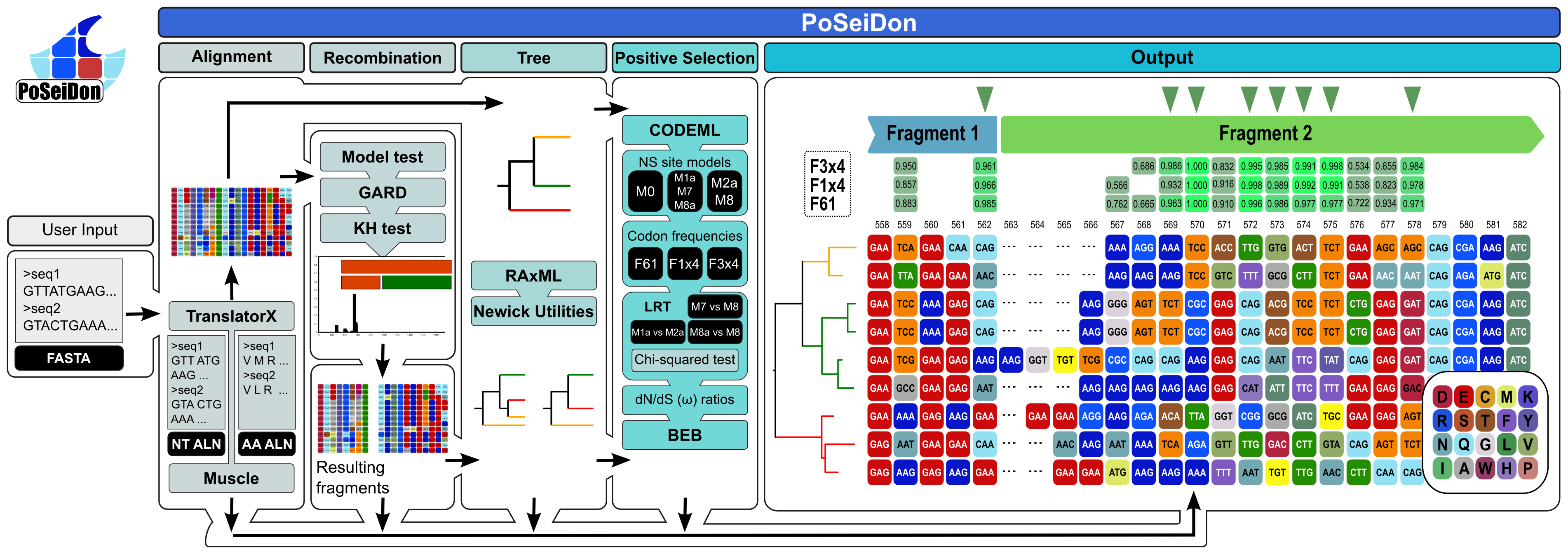
Le pipeline Poséidon comprend l'alignement dans le cadre de séquences codant pour les protéines homologues, la détection d'événements de recombinaison putatifs et les points de rupture évolutifs, les reconstructions phylogénétiques et la détection de sites sélectionnés positivement dans l'alignement complet et tous les fragments possibles. Enfin, tous les résultats sont combinés et visualisés dans une page Web HTML conviviale et claire. Les fragments d'alignement résultants sont indiqués par des barres colorées dans la sortie HTML.
TradatorX (v1.1), Abascal et al . (2010); 20435676
Muscle (v3.8.31), Edgar (2004); 15034147
RAXML (V8.0.25), Stamatakis (2014); 24451623
Newick Utilities (V1.6), Junier et Zdobnov (2010); 20472542
Modeltest, Posada et Crandall (1998); 9918953
Hyphy (v2.2), Pond et al . (2005); 15509596
Gard, Pond et al. (2006); 17110367
PAML / Codeml (V4.8), Yang (2007); 17483113
Ruby (v2.3.1)
Inkscape (V1.0)
pdftex (v3.14)
La plupart des paramètres de Poséidon sont facultatifs et sont expliqués ci-dessous en détail.
--fasta
Obligatoire. Votre fichier Fasta d'entrée doit suivre le format:
>Myotis_lucifugus Mx1 Gene ATGGCGATCGAGATACGATACGTA... >Myotis_davidii Mx1 Gene ATGGCGGTCGAGATAAGATACGTT...
Toutes les séquences doivent avoir une trame de lecture ouverte correcte, ne peuvent contenir des caractères nucléotidiques [a | c | g | t] et pas de codon d'arrêt interne.
Les ID de séquence doivent être uniques jusqu'à la première occurrence d'un espace.
--reference
Facultatif. Par défaut: utilisez l'identifiant de la première séquence comme référence. Vous pouvez définir une pièce d'identité d'espèce à partir de votre fichier Fasta multiple en tant qu'espèce de référence. Des sites sélectionnés positivement et des acides aminés correspondants seront dessinés par rapport à cette espèce. L'ID doit correspondre à l'en-tête FASTA jusqu'à l'occasion du premier espace. Par exemple, si vous voulez Myotis lucifugus comme espèce de référence et que votre fichier Fasta contient:
>Myotis_lucifugus Mx1 Gene ATGGCGATCGAGATACGATACGTA...
utiliser
--reference "Myotis_lucifugus"
comme paramètre pour définir les espèces de référence. Par défaut, le premier ID se produisant dans le fichier FastA multiple sera utilisé.
--outgroup
Facultatif. Par défaut: les arbres ne sont pas ravis. Vous pouvez définir un ou plusieurs ID d'espèces (séparées par des virgules) comme un groupe extérieur. Tous les arbres phylogénétiques seront enracinés selon cette espèce. Par exemple, si votre fichier Fasta multiple contient
ATGGCGATCGAGATACGATACGTA...
>Myotis_davidii Mx1 Gene
ATGGCGGTCGAGATAAGATACGTT...
>Pteropus_vampyrus Mx1 Gene
ATGGCCGTAGAGATTAGATACTTT...
>Eidolon_helvum Mx1 Gene
ATGCCCGTAGAGAATAGATACTTT...Vous pouvez définir:
--outgroup "Pteropus_vampyrus,Eidolon_helvum"
Pour enraciner tous les arbres par rapport à ces deux espèces.
--kh
Facultatif. Par défaut: false. Avec ce paramètre, vous pouvez décider si des points d'arrêt insignifiants doivent être pris en compte. Tous les points d'arrêt sont testés pour une incongruence topologique importante à l'aide d'un test Kashino Hasegawa (KH) Kishino, H. et Hasegawa, M. (1989). Les points d'arrêt insignifiants de KH découlent le plus souvent de la variation des longueurs de branche entre les segments. Néanmoins, prendre en compte les points d'arrêt insignifiants de KH pourrait être intéressant, car nous avons déjà observé des sites putatifs sélectionnés positivement dans des fragments sans aucune inconditionnelle topologique significative. Les fragments insignifiants de KH sont marqués dans la sortie finale, car ils pourraient ne pas se produire à partir d'événements de recombinaison réels.
Par défaut, seuls les points d'arrêt significatifs sont utilisés pour d'autres calculs.
Veuillez également garder à l'esprit que l'utilisation de points d'arrêt également insignifiants peut prolonger le temps d'exécution de Poséidon de quelques minutes en heures, selon le nombre de points d'arrêt détectés.
Veuillez consulter le --help pour d'autres paramètres (Gard, Raxml, ...) et faites-nous savoir si vous avez besoin de plus de personnalisation!
Si Poseidon vous aide, veuillez citer:
Martin Hölzer et Manja Marz, "Poséidon: un pipeline Nextflow pour la détection d'événements de recombinaison évolutive et de sélection positive", OUP Bioinformatique (2020)


