BRAKER
v3.0.8
Voici un enregistrement de la première session d'atelier BGA23 sur Braker. Si l'apprentissage en regardant des vidéos est facile pour vous, pensez à regarder cela: https://www.youtube.com/watch?v=UXTKJ4MUKYG
Braker3 est maintenant en https://usegalaxy.eu/
Tsebra & Braker3 lié:
Braker & Augustus lié:
Lié à Genemark:
Mark Borodovsky, Georgia Tech, États-Unis, [email protected]
Tomas Bruna, Joint Genome Institute, USA, [email protected]
Alexandre Lomsazde, Georgia Tech, États-Unis, [email protected]
[A] Université de Greifswald, Institute for Mathematics and Computer Science, Walther-Rathenau-Str. 47, 17489 Greifswald, Allemagne
[B] Université de Greifswald, Centre de génomique fonctionnelle des microbes, Felix-Hausdorff-Str. 8, 17489 Greifswald, Allemagne
[C] Joint Georgia Tech et Emory University Wallace H Coulter Department of Biomedical Engineering, 30332 Atlanta, USA
[D] École des sciences et de l'ingénierie informatique, 30332 Atlanta, États-Unis
[E] Moscou Institute of Physics and Technology, Moscou Region 141701, Dolgoprudny, Russie
![Braker2-Team-2 [Fig10]](https://images.downcodes.com/uploads/20250214/img_67aee79a0cb7530.png)
![Braker2-Team-1 [Fig11]](https://images.downcodes.com/uploads/20250214/img_67aee79a0d1eb31.png)
![Braker2-Team-3 [Fig12]](https://images.downcodes.com/uploads/20250214/img_67aee79a0da9c32.png)
![Braker2-Team-4 [Fig13]](https://images.downcodes.com/uploads/20250214/img_67aee79a0e49933.png)
Figure 1: Auteurs de Braker actuels, de gauche à droite: Mario Stanke, Alexandre Lomsadze, Katharina J. Hoff, Tomas Bruna, Lars Gabriel et Mark Borodovsky. Nous reconnaissons qu'une communauté plus large de scientifiques a contribué au code Braker (par exemple via les demandes de traction).
Le développement de Braker1, Braker2 et Braker3 a été soutenu par les National Institutes of Health (NIH) [GM128145 à MB et MS]. Le développement de Braker3 a été partiellement financé par la compétence des données du projet accordé à KJH et à MS par le gouvernement de Mecklenburg-Vorpommern, en Allemagne.
Le sélecteur de transcription pour Braker (Tsebra) est disponible sur https://github.com/gaius-augustus/tsebra.
Genemark-ETP, l'un des gènes au cœur de Braker, est disponible sur https://github.com/gatech-genemark/genemark-etp.
Augustus, le deuxième chercheur de gènes au cœur de Braker, est disponible sur https://github.com/gaius-augustus/augustus.
Galba, une retombée de pipeline Braker pour utiliser MiniProt ou GenomethReader pour générer des gènes d'entraînement, est disponible sur https://github.com/gaius-augustus/galba.
Le nombre en croissance rapide de génomes séquencés nécessite des méthodes entièrement automatisées pour l'annotation précise de la structure des gènes. Avec cet objectif à l'esprit, nous avons développé Braker1 R1 R0 , une combinaison de Genemark-ET R2 et Augustus R3, R4 , qui utilise des données génomiques et RNA-seq pour générer automatiquement des annotations de structure gène complète dans un nouveau génome.
Cependant, la qualité des données RNA-SEQ disponibles pour annotation d'un nouveau génome est variable, et dans certains cas, les données RNA-Seq ne sont pas du tout disponibles.
Braker2 est une extension de Braker1 qui permet une formation entièrement automatisée des outils de prédiction de gène Genemark-ES / ET / EP / ETP R14, R15, R17, F1 et Augustus à partir d'informations sur l'ARN-seq et / ou les informations sur l'homologie des protéines, et qui intègre le preuves extrinsèques des informations d'ARN-seq et d'homologie des protéines dans la prédiction .
Contrairement à d'autres méthodes disponibles qui reposent sur les informations sur l'homologie des protéines, Braker2 atteint une précision élevée de prédiction des gènes même en l'absence d'annotation d'espèces très étroitement apparentées et en l'absence de données ARN-Seq.
Braker3 est le dernier pipeline de la suite Braker. Il permet l'utilisation des données d'ARN-seq et de protéines dans un pipeline entièrement automatisé pour s'entraîner et prédire des gènes très fiables avec Genemark-ETP et Augustus. Le résultat du pipeline est l'ensemble de gènes combiné des deux outils de prédiction des gènes, qui ne contient que des gènes avec un soutien très élevé à partir de preuves extrinsèques.
Dans ce guide de l'utilisateur, nous ferons référence à Braker1, Braker2 et Braker3 simplement en tant que Braker car ils sont exécutés par le même script ( braker.pl ).
Utilisez un assemblage du génome de haute qualité. Si vous avez un grand nombre d'échafaudages très courts dans votre assemblage du génome, ces échafaudages courts augmenteront probablement considérablement le temps d'exécution mais n'augmenteront pas la précision des prédictions.
Utilisez des noms d'échafaudage simples dans le fichier du génome (par exemple >contig1 fonctionnera mieux que >contig1my custom species namesome putative function /more/information/ and lots of special characters %&!*(){} ). Rendez les noms d'échafaudage dans tous vos fichiers FastA simples avant d'exécuter n'importe quel programme d'alignement.
Afin de prédire les gènes avec précision dans un nouveau génome, le génome doit être masqué pour les répétitions. Cela évitera la prédiction des structures génétiques faussement positives dans les régions répétitives et peu complexes. Le masquage de répétition est également essentiel pour mapper les données d'ARN-Seq à un génome avec certains outils (d'autres cartographies RNA-Seq, telles que HISAT2, ignorent les informations de masquage). Dans le cas de génémemarques / ET / EP / ETP et Augustus, le masquage doux (c'est-à-dire mettre des régions répétées dans les lettres en bas de cas et toutes les autres régions dans les lettres de cas supérieure) conduit à de meilleurs résultats que le masquage dur (c'est-à-dire en remplacement des lettres dans des régions répétitives par la lettre N pour les nucléotides inconnus).
De nombreux génomes ont des structures génétiques qui seront prédites avec précision avec les paramètres standard des génémers / ET / EP / ETP et Augustus au sein de Braker. Cependant, certains génomes ont des caractéristiques spécifiques au clade, c'est-à-dire un modèle de point de branche spécial dans des champignons ou des motifs d'épissage non standard. Veuillez lire la section des options [Options] afin de déterminer si l'une des options personnalisées peut améliorer la précision de la prédiction des gènes dans le génome de votre espèce cible.
Vérifiez toujours les résultats de la prédiction des gènes avant de poursuivre l'utilisation! Vous pouvez utiliser un navigateur de génome pour l'inspection visuelle des modèles de gènes en contexte avec des données de preuves extrinsèques. Braker soutient la génération de pôles de données de piste pour le navigateur du génome UCSC avec Makehub à cet effet.
Braker propose principalement des données de preuves extrinsèques semi-instratisées (informations d'alignement épissées sur l'ARN-seq et / ou en protéines), la formation soutenue de Genemark-ES / ET / EP / ETP [F1] et la formation ultérieure de l'Augustus avec l'intégration des preuves extrinsèques dans la finale étape de prédiction des gènes. Cependant, il y a maintenant un certain nombre de pipelines supplémentaires inclus dans Braker. Dans ce qui suit, nous donnons un aperçu des fichiers d'entrée possibles et des pipelines:
![Braker2-main-a [Fig1]](https://images.downcodes.com/uploads/20250214/img_67aee79a0eaf534.png)
Figure 2: Braker Pipeline A: formation de génériques-ES sur les données du génome, seulement; AB Initio Gene Prediction withaugustus
![Braker2-main-b [Fig2]](https://images.downcodes.com/uploads/20250214/img_67aee79a0f13f35.png)
Figure 3: Braker Pipeline B: Formation Genemark-ET soutenue par des informations d'alignement épissées RNA-SEQ, prédiction avec Auguste avec ces mêmes informations d'alignement épissées.
![Braker2-Main-C [Fig3]](https://images.downcodes.com/uploads/20250214/img_67aee79a0fa1036.png)
Figure 4: Braker Pipeline C: formation Genemark-EP + sur l'alignement épissé des protéines, le démarrage et l'arrêt des informations, la prédiction avec Auguste avec les mêmes informations, en plus des indices CDSPART enchaînés. Les protéines utilisées ici peuvent être de toute distance évolutive à l'organisme cible.
![Braker3-main-a [Fig4]](https://images.downcodes.com/uploads/20250214/img_67aee79a1010b37.png)
Figure 5: Braker Pipeline D: Si nécessaire, téléchargez et alignement des ensembles d'ARN-seq pour les espèces cibles. La formation de Genemark-ETP soutenue par les alignements RNA-Seq et une grande base de données de protéines (les protéines peuvent être de n'importe quelle distance évolutive). Par la suite, la formation et la prédiction d'Auguste en utilisant les mêmes informations extrinsèques avec les résultats Genemark-ETP. La prédiction finale est la combinaison Tsebra des résultats Auguste et Genemark-ETP.
Nous sommes conscients que l'installation "manuelle" de Braker3 et de toutes ses dépendances est fastidieuse et vraiment difficile sans autorisation radiculaire. Par conséquent, nous fournissons un conteneur Docker qui a été développé pour être géré avec la singularité. Toutes les informations sur ce conteneur se trouvent sur https://hub.docker.com/r/teambraker/braker3
Bref, construisez-le comme suit:
singularity build braker3.sif docker://teambraker/braker3:latest
Exécuter avec:
singularity exec braker3.sif braker.pl
Tester avec:
singularity exec -B $PWD:$PWD braker3.sif cp /opt/BRAKER/example/singularity-tests/test1.sh .
singularity exec -B $PWD:$PWD braker3.sif cp /opt/BRAKER/example/singularity-tests/test2.sh .
singularity exec -B $PWD:$PWD braker3.sif cp /opt/BRAKER/example/singularity-tests/test3.sh .
export BRAKER_SIF=/your/path/to/braker3.sif # may need to modify
bash test1.sh
bash test2.sh
bash test3.sh
Peu d'utilisateurs souhaitent exécuter leur analyse dans Docker (car les autorisations racines sont nécessaires). Cependant, si c'est votre objectif, vous pouvez courir et tester le conteneur comme suit
sudo docker run --user 1000:100 --rm -it teambraker/braker3:latest bash
bash /opt/BRAKER/example/docker-tests/test1.sh # BRAKER1
bash /opt/BRAKER/example/docker-tests/test2.sh # BRAKER2
bash /opt/BRAKER/example/docker-tests/test3.sh # BRAKER3
Bonne chance ;-)
$PATH pourraient conduire à des interférences imprévues, provoquant des échecs de programme. Veuillez déplacer toutes les versions Genemark plus anciennes de votre $PATH (par exemple, par exemple le Genemark en ProtHint/dependencies ).
Au moment de la sortie, cette version Braker a été testée avec:
Auguste 3.5.0 F2
Genemark-ETP (Source Voir Dockerfile)
Bamtools 2.5.1 R5
SAMTOOLS 1.7-4-G93586ED R6
Spaln 2.3.3d R8, R9, R10
NCBI BLAST + 2.2.31+ R12, R13
Diamant 0.9.24
cdbfasta 0.99
CDBYANK 0.981
GUSHR 1.0.0
SRA Toolkit 3.00 R14
HISAT2 2.2.1 R15
Bedtools 2.30 R16
Stringtie2 2.2.1 R17
Gffread 0.12,7 R18
complexe 0,2,5 R27
La course à pied nécessite un système linux avec bash et perl. De plus, Braker nécessite l'installation des modules CPAN-PERL suivants:
File::Spec::Functions
Hash::Merge
List::Util
MCE::Mutex
Module::Load::Conditional
Parallel::ForkManager
POSIX
Scalar::Util::Numeric
YAML
Math::Utils
File::HomeDir
Pour Genemark-ETP, utilisé lorsque la protéine et l'ARN-Seq sont fournis:
YAML::XSData::DumperThread::Queuethreads Sur Ubuntu, par exemple, installez les modules avec CPANMINUS F4 : sudo cpanm Module::Name , par exemple sudo cpanm Hash::Merge .
Braker utilise également un module Perl helpMod_braker.pm qui n'est pas disponible sur CPAN. Ce module fait partie de la version Braker et ne nécessite pas d'installation distincte.
Si vous n'avez pas d'autorisations racine sur la machine Linux, essayez de configurer un environnement Anaconda (https://www.anaconda.com/distribution/) comme suit:
wget https://repo.anaconda.com/archive/Anaconda3-2018.12-Linux-x86_64.sh
bash bin/Anaconda3-2018.12-Linux-x86_64.sh # do not install VS (needs root privileges)
conda install -c anaconda perl
conda install -c anaconda biopython
conda install -c bioconda perl-app-cpanminus
conda install -c bioconda perl-file-spec
conda install -c bioconda perl-hash-merge
conda install -c bioconda perl-list-util
conda install -c bioconda perl-module-load-conditional
conda install -c bioconda perl-posix
conda install -c bioconda perl-file-homedir
conda install -c bioconda perl-parallel-forkmanager
conda install -c bioconda perl-scalar-util-numeric
conda install -c bioconda perl-yaml
conda install -c bioconda perl-class-data-inheritable
conda install -c bioconda perl-exception-class
conda install -c bioconda perl-test-pod
conda install -c bioconda perl-file-which # skip if you are not comparing to reference annotation
conda install -c bioconda perl-mce
conda install -c bioconda perl-threaded
conda install -c bioconda perl-list-util
conda install -c bioconda perl-math-utils
conda install -c bioconda cdbtools
conda install -c eumetsat perl-yaml-xs
conda install -c bioconda perl-data-dumper
Installez par la suite Braker et d'autres logiciels "comme d'habitude" tout en étant dans votre environnement Conda. Remarque: Il y a un package Bioconda Braker et un package Bioconda Augustus. Ils fonctionnent. Mais ils sont généralement à la traîne du code de développement des deux outils sur GitHub. Nous recommandons donc l'installation manuelle et l'utilisation de dernières sources.
Braker est une collection de scripts Perl et Python et un module Perl. Le script principal qui sera appelé pour exécuter Braker est braker.pl . Les composants Perl et Python supplémentaires sont:
align2hints.pl
filterGenemark.pl
filterIntronsFindStrand.pl
startAlign.pl
helpMod_braker.pm
findGenesInIntrons.pl
downsample_traingenes.pl
ensure_n_training_genes.py
get_gc_content.py
get_etp_hints.py
Tous les scripts (fichiers se terminant par *.pl et *.py ) qui font partie de Braker doivent être exécutables pour exécuter Braker. Cela devrait déjà être le cas si vous téléchargez Braker de GitHub. L'exécutabilité peut être écrasée si vous transférez Braker sur un bricolage USB vers un autre ordinateur. Afin de vérifier si les fichiers requis sont exécutables, exécutez la commande suivante dans le répertoire contenant des scripts Braker Perl:
ls -l *.pl *.py
La sortie doit être similaire à ceci:
-rwxr-xr-x 1 katharina katharina 18191 Mai 7 10:25 align2hints.pl
-rwxr-xr-x 1 katharina katharina 6090 Feb 19 09:35 braker_cleanup.pl
-rwxr-xr-x 1 katharina katharina 408782 Aug 17 18:24 braker.pl
-rwxr-xr-x 1 katharina katharina 5024 Mai 7 10:25 downsample_traingenes.pl
-rwxr-xr-x 1 katharina katharina 5024 Mai 7 10:23 ensure_n_training_genes.py
-rwxr-xr-x 1 katharina katharina 4542 Apr 3 2019 filter_augustus_gff.pl
-rwxr-xr-x 1 katharina katharina 30453 Mai 7 10:25 filterGenemark.pl
-rwxr-xr-x 1 katharina katharina 5754 Mai 7 10:25 filterIntronsFindStrand.pl
-rwxr-xr-x 1 katharina katharina 7765 Mai 7 10:25 findGenesInIntrons.pl
-rwxr-xr-x 1 katharina katharina 1664 Feb 12 2019 gatech_pmp2hints.pl
-rwxr-xr-x 1 katharina katharina 2250 Jan 9 13:55 log_reg_prothints.pl
-rwxr-xr-x 1 katharina katharina 4679 Jan 9 13:55 merge_transcript_sets.pl
-rwxr-xr-x 1 katharina katharina 41674 Mai 7 10:25 startAlign.pl
Il est important que le x in -rwxr-xr-x soit présent pour chaque script. Si ce n'est pas le cas, courez
`chmod a+x *.pl *.py`
Afin de modifier les attributs de fichiers.
Vous pouvez trouver utile d'ajouter le répertoire dans lequel les scripts Braker Perl résident à votre variable d'environnement $PATH . Pour une seule session bash, entrez:
PATH=/your_path_to_braker/:$PATH
export PATH
Pour rendre cette modification $PATH à la disposition de toutes les sessions de bash, ajoutez les lignes ci-dessus à un script de démarrage (par exemple ~/.bashrc ).
Braker fait appel à divers outils logiciels bioinformatiques qui ne font pas partie de Braker. Certains outils sont obligatoires, c'est-à-dire que Braker ne fonctionnera pas du tout si ces outils ne sont pas présents sur votre système. Les autres outils sont facultatifs. Veuillez installer tous les outils requis pour exécuter Braker dans le mode de votre choix.
Téléchargez Genemark-etp F1 à partir de http://github.com/gatech-genemark/genemark-etp ou https://topaz.gatech.edu/genemark/etp.for_braker.tar.gz. Déballer et installer Genemark-ETP comme décrit dans le fichier README de GeneMark-ETP.
Si vous êtes déjà contenu dans votre variable $PATH , Braker devinera automatiquement l'emplacement de gmes_petap.pl ou gmetp.pl . Sinon, Braker peut trouver des exécutables GeneMark-ES / Et / EP / ETP en les localisant dans une variable d'environnement GENEMARK_PATH , soit en prenant un argument de ligne de commande ( --GENEMARK_PATH=/your_path_to_GeneMark_executables/ ).
Afin de définir la variable d'environnement pour votre session de bash actuelle, Type:
export GENEMARK_PATH=/your_path_to_GeneMark_executables/
Ajoutez les lignes ci-dessus à un script de démarrage (par exemple ~/.bashrc ) afin de la mettre à la disposition de toutes les sessions de bash.
Les scripts Perl dans Genemark-ES / Et / EP / ETP sont configurés avec l'emplacement Perl par défaut à /usr/bin/perl .
Si vous exécutez des génémers / eT / EP / ETP dans un environnement Anaconda (ou souhaitez utiliser Perl à partir de la variable $PATH pour toute autre raison), modifiez le shebang de tous les scripts génémers / ET / EP / ETP avec La commande suivante située à l'intérieur du dossier Genemark-ES / ET / EP / ETP:
perl change_path_in_perl_scripts.pl "/usr/bin/env perl"
Vous pouvez vérifier si Genemark-ES / ET / EP est installé correctement en exécutant le check_install.bash et / ou en exécutant des exemples dans le répertoire GeneMark-E-tests .
Genemark-ETP est compatible à la baisse, c'est-à-dire qu'il couvre également la fonctionnalité de Genemark-EP et Genemark-ET dans Braker.
Téléchargez Augustus depuis sa branche maître à https://github.com/gaius-augustus/augustus. Déballer Augustus et installer Augustus selon Augustus README.TXT . N'utilisez pas les versions Augustus obsolètes provenant d'autres sources, par exemple, le package Debian ou le package Bioconda! Braker dépend fortement en particulier d'un répertoire Auguste / Scripts à jour, et d'autres sources sont souvent en retard.
Vous devez compiler Augustus sur votre propre système afin d'éviter les problèmes de versions des bibliothèques utilisées par Augustus. Des instructions de compilation sont fournies dans le fichier Augustus README.TXT ( Augustus/README.txt ).
Augustus se compose d' augustus , de l'outil de prédiction des gènes, d'outils C ++ supplémentaires situés dans Augustus/auxprogs et Perl Scripts situés dans Augustus/scripts . Les scripts Perl doivent être exécutables (voir les instructions dans les composants de la section Braker.
L'outil C ++ bam2hints est un composant essentiel de Braker lorsqu'il est exécuté avec RNA-seq. Des sources sont situées dans Augustus/auxprogs/bam2hints . Assurez-vous de compiler bam2hints sur votre système (il doit être automatiquement compilé lorsque Augustus est compilé, mais en cas de problèmes avec bam2hints , veuillez lire les instructions de dépannage dans Augustus/auxprogs/bam2hints/README ).
Étant donné que Braker est un pipeline qui forme Augustus, IE écrit des fichiers de paramètres spécifiques aux espèces, Braker a besoin d'écrire un accès au répertoire de configuration d'Augustus qui contient de tels fichiers ( Augustus/config/ ). Si vous installez Augustus globalement sur votre système, le dossier config ne sera généralement pas écrit par tous les utilisateurs. Soit faire le répertoire où config réside récursivement écrit aux utilisateurs d'Augustus, soit copier le dossier config/ (récursivement) à un emplacement où les utilisateurs ont une autorisation d'écriture.
Augustus localisera le dossier config en recherchant une variable d'environnement $AUGUSTUS_CONFIG_PATH . Si la variable d'environnement $AUGUSTUS_CONFIG_PATH n'est pas définie, Braker examinera le chemin ../config par rapport au répertoire dans lequel il trouve un exécutable Augustus. Alternativement, vous pouvez fournir la variable en tant qu'argument de ligne de commande à Braker ( --AUGUSTUS_CONFIG_PATH=/your_path_to_AUGUSTUS/Augustus/config/ ). Nous vous recommandons d'exporter la variable, par exemple pour votre session de bash actuelle:
export AUGUSTUS_CONFIG_PATH=/your_path_to_AUGUSTUS/Augustus/config/
Afin de mettre la variable à la disposition de toutes les sessions de bash, ajoutez la ligne ci-dessus à un script de démarrage, par exemple ~/.bashrc .
Veuillez consulter le Dockerfile au cas où vous souhaitez installer Augustus en tant que package Debian. Un certain nombre de scripts doivent donc être corrigés.
Braker attend l'intégralité du répertoire config d'Augustus à $AUGUSTUS_CONFIG_PATH , c'est-à-dire les species de sous-dossiers avec son contenu (au moins generic ) et extrinsic ! Fournir un dossier écrit mais vide à $AUGUSTUS_CONFIG_PATH ne fonctionnera pas pour Braker. Si vous devez séparer Augustus Binary et $AUGUSTUS_CONFIG_PATH , nous vous recommandons de copier de manière récursive le contenu de configuration non-réparable à un emplacement écrivatif.
Si vous avez une installation à l'échelle du système d'Augustus à /usr/bin/augustus , une copie non réactible de config se trouve à /usr/bin/augustus_config/ . Le dossier /home/yours/ vous est écrit. Copiez avec la commande suivante (et définissez en outre les variables alors requises):
cp -r /usr/bin/Augustus/config/ /home/yours/
export AUGUSTUS_CONFIG_PATH=/home/yours/augustus_config
export AUGUSTUS_BIN_PATH=/usr/bin
export AUGUSTUS_SCRIPTS_PATH=/usr/bin/augustus_scripts
L'ajout de répertoires de binaires et de scripts Augustus à votre variable $PATH permet à votre système de localiser ces outils, automatiquement. Il n'est pas exigé de faire fonctionner Braker de le faire, car Braker essaiera de les deviner à partir de l'emplacement d'une autre variable d'environnement ( $AUGUSTUS_CONFIG_PATH ), ou les deux répertoires peuvent être fournis comme arguments de ligne de commande à braker.pl , mais nous recommandons de Ajoutez-les à votre variable $PATH . Pour votre session de bash actuelle, Type:
PATH=:/your_path_to_augustus/bin/:/your_path_to_augustus/scripts/:$PATH
export PATH
Pour toutes vos sessions bash, ajoutez les lignes ci-dessus à un script de démarrage (par exemple ~/.bashrc ).
Sur Ubuntu, Python3 est généralement installé par défaut, python3 sera dans votre variable $PATH , par défaut, et Braker le localisera automatiquement. Cependant, vous avez la possibilité de spécifier l'emplacement binaire python3 de deux autres manières:
Exporter une variable d'environnement $PYTHON3_PATH , par exemple dans votre fichier ~/.bashrc :
export PYTHON3_PATH=/path/to/python3/
Spécifiez l'option de ligne de commande --PYTHON3_PATH=/path/to/python3/ vers braker.pl .
Téléchargez Bamtools (par exemple git clone https://github.com/pezmaster31/bamtools.git ). Installez Bamtools en tapant ce qui suit dans votre coquille:
cd your-bamtools-directory mkdir build cd build cmake .. make
Si déjà dans votre variable $PATH , Braker trouvera automatiquement Bamtools. Sinon, Braker peut localiser le binaire Bamtools en utilisant une variable d'environnement $BAMTOOLS_PATH , soit en prenant un argument de ligne de commande ( --BAMTOOLS_PATH=/your_path_to_bamtools/bin/ f6 ). Afin de définir la variable d'environnement, par exemple pour votre session de bash actuelle, Type:
export BAMTOOLS_PATH=/your_path_to_bamtools/bin/
Ajoutez la ligne ci-dessus à un script de démarrage (par exemple ~/.bashrc ) afin de définir la variable d'environnement pour toutes les sessions bash.
Vous pouvez utiliser NCBI Blast + ou Diamond pour l'élimination des gènes d'entraînement redondants. Vous n'avez pas besoin des deux outils. Si le diamant est présent, il sera préféré car il est beaucoup plus rapide.
Obtenez et déballer le diamant comme suit:
wget http://github.com/bbuchfink/diamond/releases/download/v0.9.24/diamond-linux64.tar.gz
tar xzf diamond-linux64.tar.gz
Si déjà dans votre variable $PATH , Braker trouvera automatiquement Diamond. Sinon, Braker peut localiser le binaire Diamond en utilisant une variable d'environnement $DIAMOND_PATH , soit en prenant un argument de ligne de commande ( --DIAMOND_PATH=/your_path_to_diamond ). Afin de définir la variable d'environnement, par exemple pour votre session de bash actuelle, Type:
export DIAMOND_PATH=/your_path_to_diamond/
Ajoutez la ligne ci-dessus à un script de démarrage (par exemple ~/.bashrc ) afin de définir la variable d'environnement pour toutes les sessions bash.
Si vous décidez de BLAST +, installez NCBI BLAST + avec sudo apt-get install ncbi-blast+ .
Si déjà dans votre variable $PATH , Braker trouvera automatiquement BLASTP. Sinon, Braker peut localiser le binaire BLASTP soit en utilisant une variable d'environnement $BLAST_PATH , soit en prenant un argument de ligne de commande ( --BLAST_PATH=/your_path_to_blast/ ). Afin de définir la variable d'environnement, par exemple pour votre session de bash actuelle, Type:
export BLAST_PATH=/your_path_to_blast/
Ajoutez la ligne ci-dessus à un script de démarrage (par exemple ~/.bashrc ) afin de définir la variable d'environnement pour toutes les sessions bash.
Les outils suivants sont requis par GeneMark-ETP et il essaiera de les localiser dans votre variable $PATH . Assurez-vous donc d'ajouter leur emplacement à votre $PATH , par exemple:
export PATH=$PATH:/your/path/to/Tool
Pour tous les outils ci-dessous, ajoutez la ligne ci-dessus à un script de démarrage (par exemple ~/.bashrc ) afin d'étendre votre variable $PATH pour toutes les sessions de bash.
Ces outils logiciels ne sont obligatoires que si vous exécutez Braker avec des données RNA-Seq et Protein!
StringTie2 est utilisé par Genemark-ETP pour assembler les alignements ARN-seq alignés. Une version précompilée de Stringtie2 peut être téléchargée à partir de https://ccb.jhu.edu/software/stringtie/#install.
Le package logiciel BedTools est requis par Genemark-ETP si vous souhaitez exécuter Braker avec les données RNA-Seq et Protein. Vous pouvez télécharger BedTools à partir de https://github.com/arq5x/bedtools2/releases. Ici, vous pouvez soit télécharger une version précompilée bedtools.static.binary , par exemple
wget https://github.com/arq5x/bedtools2/releases/download/v2.30.0/bedtools.static.binary
mv bedtools.static.binary bedtools
chmod a+x
ou vous pouvez télécharger bedtools-2.30.0.tar.gz et le compiler à partir de la source en utilisant make , par exemple
wget https://github.com/arq5x/bedtools2/releases/download/v2.30.0/bedtools-2.30.0.tar.gz
tar -zxvf bedtools-2.30.0.tar.gz
cd bedtools2
make
Voir https://bedtools.readthedocs.io/en/latest/content/installation.html pour plus d'informations.
Gffread est un logiciel d'utilité requis par Genemark-ETP. Il peut être téléchargé à partir de https://github.com/gpertea/gffread/releases/download/v0.12.7/gffread-0.12.7.linux_x86_64.tar.gz et installé avec make , par exemple
wget https://github.com/gpertea/gffread/releases/download/v0.12.7/gffread-0.12.7.Linux_x86_64.tar.gz
tar xzf gffread-0.12.7.Linux_x86_64.tar.gz
cd gffread-0.12.7.Linux_x86_64
make
Samtools n'est pas requis pour exécuter Braker sans GeneMark-ETP si tous vos fichiers sont formatés, correctement (c'est-à-dire que toutes les séquences doivent avoir des noms Fasta courts et uniques). Si vous n'êtes pas sûr de savoir si tous vos fichiers sont correctement fomatés, il peut être utile d'installer SAMTools car Braker peut automatiquement résoudre certains problèmes de format en utilisant SAMTools.
Comme condition préalable à Samtools, téléchargez et installez htslib (par exemple git clone https://github.com/samtools/htslib.git , suivez la documentation htslib pour l'installation).
Téléchargez et installez Samtools (par exemple git clone git://github.com/samtools/samtools.git ), suivez ensuite la documentation SAMTools pour l'installation).
Si déjà dans votre variable $PATH , Braker trouvera automatiquement Samtools. Sinon, Braker peut trouver Samtools soit en prenant un argument de ligne de commande ( --SAMTOOLS_PATH=/your_path_to_samtools/ ), ou en utilisant une variable d'environnement $SAMTOOLS_PATH . Pour exporter la variable, par exemple pour votre session de bash actuelle, Type:
export SAMTOOLS_PATH=/your_path_to_samtools/
Ajoutez la ligne ci-dessus à un script de démarrage (par exemple ~/.bashrc ) afin de définir la variable d'environnement pour toutes les sessions bash.
Si Biopython est installé, Braker peut générer des Fasta-Files avec des séquences de codage et des séquences de protéines prédites par Augustus et générer des pôles de données de piste pour la visualisation d'un Braker exécuté avec Makehub R16 . Ce sont des étapes facultatives. Le premier peut être désactivé avec l'indicateur de ligne de commande --skipGetAnnoFromFasta , le second peut être activé en utilisant les options de ligne de commande --makehub [email protected] , Biopython n'est pas requis si aucune de ces étapes facultatives facultatives doit être effectué.
Sur Ubuntu, installez Python3 Package Manager avec:
`sudo apt-get install python3-pip`
Ensuite, installez Biopython avec:
`sudo pip3 install biopython`
CDBFasta et CdByank sont requis par Braker pour corriger les gènes Augustus avec des codons d'arrêt de trame (codons d'arrêt épissés) à l'aide du script Augustus fix_in_frame_stop_codon_genes.py. Cela peut être ignoré avec --skip_fixing_broken_genes .
Sur Ubuntu, installez CDBFasta avec:
sudo apt-get install cdbfasta
Pour d'autres systèmes, vous pouvez par exemple obtenir CDBFasta à partir de https://github.com/gpertea/cdbfasta, par exemple:
git clone https://github.com/gpertea/cdbfasta.git
cd cdbfasta
make all
Sur Ubuntu, CDBFasta et CDBYank seront dans votre variable $PATH après l'installation, et Braker les localisera automatiquement. Cependant, vous avez la possibilité de spécifier l'emplacement binaire cdbfasta et cdbyank de deux autres manières:
$CDBTOOLS_PATH , par exemple dans votre fichier ~/.bashrc : export CDBTOOLS_PATH=/path/to/cdbtools/
--CDBTOOLS_PATH=/path/to/cdbtools/ vers braker.pl . Remarque: Le soutien de la Spaln autonome (ouside de Prhint) dans Braker est obsolète.
Cet outil est requis si vous exécutez Prhint ou si vous souhaitez exécuter des alignements de protéines au génome avec Braker en utilisant Spaln en dehors de Prhint. L'utilisation de Spaln en dehors de Prhint est une approche appropriée que si une espèce annotée de courte distance évolutive à votre génome cible est disponible. Nous vous recommandons de diriger Spaln via Prhint pour Braker. Pruint apporte un binaire Spaln. Si cela ne fonctionne pas sur votre système, téléchargez Spaln depuis https://github.com/ogotoh/spaln. Déballer et installer selon spaln/doc/SpalnReadMe22.pdf .
Braker essaiera de localiser l'exécutable Spaln en utilisant une variable d'environnement $ALIGNMENT_TOOL_PATH . Alternativement, cela peut être fourni comme argument de ligne de commande ( --ALIGNMENT_TOOL_PATH=/your/path/to/spaln ).
Cet outil n'est requis que si vous souhaitez ajouter des UTR (des données RNA-Seq) aux gènes prédits ou si vous souhaitez former des paramètres UTR pour Auguste et prédire les gènes avec des UTR. Dans tous les cas, GUSHR nécessite l'entrée des données RNA-Seq.
Gushr est disponible en téléchargement sur https://github.com/gaius-augustus/gushr. Obtenez-le en tapant:
git clone https://github.com/Gaius-Augustus/GUSHR.git
GUSHR exécute un fichier JAR Gemoma R19, R20, R21 , et ce fichier JAR nécessite Java 1.8. Sur Ubuntu, vous pouvez installer Java 1.8 avec la commande suivante:
sudo apt-get install openjdk-8-jdk
Si plusieurs versions Java sont installées sur votre système, assurez-vous d'activer 1,8 Braker en cours d'exécution avec Java en fonctionnant
sudo update-alternatives --config java
et sélectionner la version correcte.
Si vous changez --UTR=on , bamtowig.py nécessitera les outils suivants qui peuvent être téléchargés à partir de http://hgdownload.soe.ucsc.edu/admin/exe:
twobitinfo
fatotwobit
Il est facultatif d'installer ces outils dans votre chemin $. Si vous ne le faites pas et que vous changez --UTR=on , bamtowig.py les téléchargera automatiquement dans le répertoire de travail.
Si vous souhaitez générer automatiquement un centre de données de piste de votre exécution Braker, le logiciel Makehub, disponible sur https://github.com/gaius-augustus/makehub est requis. Téléchargez le logiciel (soit en exécutant git clone https://github.com/Gaius-Augustus/MakeHub.git , soit en choisissant une version à partir de https://github.com/gaius-augustus/makehub/releases. Package si vous avez téléchargé une version (par exemple, unzip MakeHub.zip ou tar -zxvf MakeHub.tar.gz .
Braker essaiera de localiser le script Make_Hub.py en utilisant une variable d'environnement $MAKEHUB_PATH . Alternativement, cela peut être fourni comme argument de ligne de commande ( --MAKEHUB_PATH=/your/path/to/MakeHub/ ). Braker peut également essayer de deviner l'emplacement de Makehub sur votre système.
Si vous voulez que Braker télécharge les bibliothèques RNA-Seq à partir de SRA de NCBI, la boîte à outils SRA est requise. Vous pouvez obtenir une version précompilée de la boîte à outils SRA à partir de http://daehwankimlab.github.io/hisat2/download/#version-hisat2-221.
Braker essaiera de trouver des binaires exécutables à partir de la boîte à outils SRA (FastQ-Dump, Prefetch) en utilisant une variable d'environnement $SRATOOLS_PATH . Alternativement, cela peut être fourni comme argument de ligne de commande ( --SRATOOLS_PATH=/your/path/to/SRAToolkit/ ). Braker peut également essayer de deviner l'emplacement de la boîte à outils SRA sur votre système si les exécutables sont dans votre variable $PATH .
Si vous souhaitez utiliser des lectures d'ARN-seq non alignées, le logiciel HISAT2 est nécessaire pour les cartographier au génome. Une version précompilée de HISAT2 peut être téléchargée à partir de http://daehwankimlab.github.io/hisat2/download/#version-hisat2-221.
Braker essaiera de trouver des binaires Hisat2 exécutables (HISAT2, HISAT2-BUILD) en utilisant une variable d'environnement $HISAT2_PATH . Alternativement, cela peut être fourni comme argument de ligne de commande ( --HISAT2_PATH=/your/path/to/HISAT2/ ). Braker peut également essayer de deviner l'emplacement de HISAT2 sur votre système si les exécutables sont dans votre variable $PATH .
Si vous souhaitez exécuter Tsebra dans Braker dans un mode de maximisation de l'exhaustivité de Busco, vous devez installer Complementsm.
wget https://github.com/huangnengCSU/compleasm/releases/download/v0.2.4/compleasm-0.2.4_x64-linux.tar.bz2
tar -xvjf compleasm-0.2.4_x64-linux.tar.bz2 &&
Ajoutez le dossier résultant compleasm_kit à votre variable $PATH , par exemple:
export PATH=$PATH:/your/path/to/compleasm_kit
Compleasm nécessite des pandas, qui peuvent être installés avec:
pip install pandas
Braker (Braker.pl) utilise GetConf pour voir combien de threads peuvent être exécutés sur votre système. Sur Ubuntu, vous pouvez l'installer avec:
sudo apt-get install libc-bin
Dans ce qui suit, nous décrivons que Braker «typique» appelle différents types de données d'entrée. En général, nous vous recommandons d'exécuter Braker sur des séquences génomiques qui ont été en masquage à la manière des répétitions. Braker ne doit être appliqué qu'à des génomes qui ont été en masquage à Soft pour les répétitions!
This approach is suitable for genomes of species for which RNA-Seq libraries with good transcriptome coverage are available and for which protein data is not at hand. The pipeline is illustrated in Figure 2.
BRAKER has several ways to receive RNA-Seq data as input:
You can provide ID(s) of RNA-Seq libraries from SRA (in case of multiple IDs, separate them by comma) as argument to --rnaseq_sets_ids . The libraries belonging to the IDs are then downloaded automatically by BRAKER, eg:
braker.pl --species=yourSpecies --genome=genome.fasta
--rnaseq_sets_ids=SRA_ID1,SRA_ID2
You can use local FASTQ file(s) of unaligned reads as input. In this case, you have to provide BRAKER with the ID(s) of the RNA-Seq set(s) as argument to --rnaseq_sets_ids and the path(s) to the directories, where the FASTQ files are located as argument to --rnaseq_sets_dirs . For each ID ID , BRAKER will search in these directories for one FASTQ file named ID.fastq if the reads are unpaired, or for two FASTQ files named ID_1.fastq and ID_2.fastq if they are paired.
For example, if you have a paired library called 'SRA_ID1' and an unpaired library named 'SRA_ID2', you have to have a directory /path/to/local/fastq/files/ , where the files SRA_ID1_1.fastq , SRA_ID1_2.fastq , and SRA_ID2.fastq reside. Then, you could run BRAKER with following command:
braker.pl --species=yourSpecies --genome=genome.fasta
--rnaseq_sets_ids=SRA_ID1,SRA_ID2
--rnaseq_sets_dirs=/path/to/local/fastq/files/
There are two ways of supplying BRAKER with RNA-Seq data as bam file(s). First, you can do it in the same way as you would supply FASTQ file(s): Provide the ID(s)/name(s) of your bam file(s) as argument to --rnaseq_sets_ids and specify directories where the bam files reside with --rnaseq_sets_dirs . BRAKER will automatically detect that these ID(s) are bam and not FASTQ file(s), eg:
braker.pl --species=yourSpecies --genome=genome.fasta
--rnaseq_sets_ids=BAM_ID1,BAM_ID2
--rnaseq_sets_dirs=/path/to/local/bam/files/
Second, you can specify the paths to your bam file(s) directly, eg can either extract RNA-Seq spliced alignment information from bam files, or it can use such extracted information, directly.
braker.pl --species=yourSpecies --genome=genome.fasta
--bam=file1.bam,file2.bam
Please note that we generally assume that bam files were generated with HiSat2 because that is the aligner that would also be executed by BRAKER3 with fastq input. If you want for some reason to generate the bam files with STAR, use the option --outSAMstrandField intronMotif of STAR to produce files that are compatible wiht StringTie in BRAKER3.
In order to run BRAKER with RNA-Seq spliced alignment information that has already been extracted, run:
braker.pl --species=yourSpecies --genome=genome.fasta
--hints=hints1.gff,hints2.gff
The format of such a hints file must be as follows (tabulator separated file):
chrName b2h intron 6591 8003 1 + . pri=4;src=E
chrName b2h intron 6136 9084 11 + . mult=11;pri=4;src=E
...
The source b2h in the second column and the source tag src=E in the last column are essential for BRAKER to determine whether a hint has been generated from RNA-Seq data.
It is also possible to provide RNA-Seq sets in different ways for the same BRAKER run, any combination of above options is possible. It is not recommended to provide RNA-Seq data with --hints if you run BRAKER in ETPmode (RNA-Seq and protein data), because GeneMark-ETP won't use these hints!
This approach is suitable for genomes of species for which no RNA-Seq libraries are available. A large database of proteins (with possibly longer evolutionary distance to the target species) should be used in this case. This mode is illustrated in figure 9.
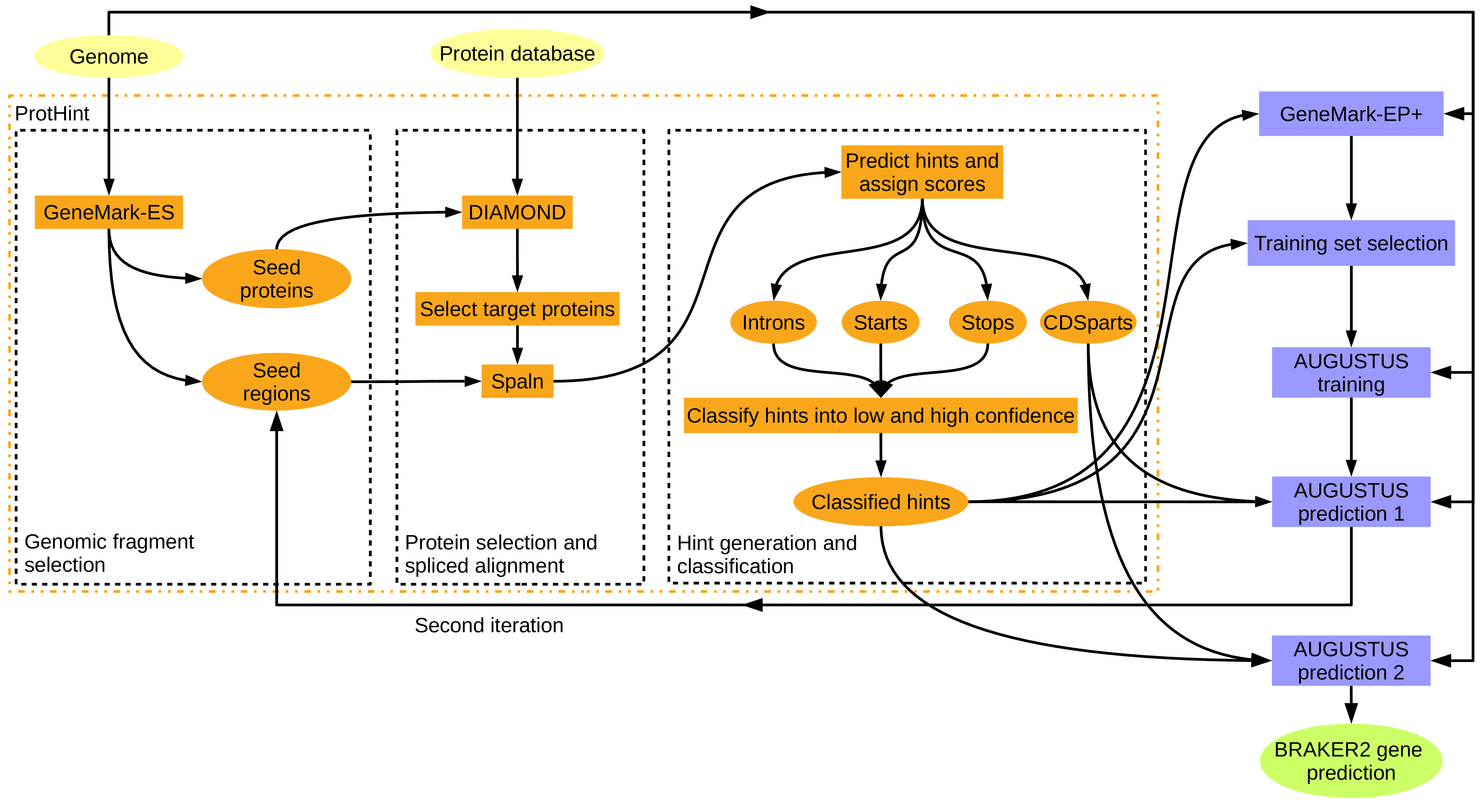
Figure 9: BRAKER with proteins of any evolutionary distance. ProtHint protein mapping pipelines is used to generate protein hints. ProtHint automatically determines which alignments are from close relatives, and which are from rather distant relatives.
For running BRAKER in this mode, type:
braker.pl --genome=genome.fa --prot_seq=proteins.fa
We recommend using OrthoDB as basis for proteins.fa . The instructions on how to prepare the input OrthoDB proteins are documented here: https://github.com/gatech-genemark/ProtHint#protein-database-preparation.
You can of course add additional protein sequences to that file, or try with a completely different database. Any database will need several representatives for each protein, though.
Instead of having BRAKER run ProtHint, you can also start BRAKER with hints already produced by ProtHint, by providing ProtHint's prothint_augustus.gff output:
braker.pl --genome=genome.fa --hints=prothint_augustus.gff
The format of prothint_augustus.gff in this mode looks like this:
2R ProtHint intron 11506230 11506648 4 + . src=M;mult=4;pri=4
2R ProtHint intron 9563406 9563473 1 + . grp=69004_0:001de1_702_g;src=C;pri=4;
2R ProtHint intron 8446312 8446371 1 + . grp=43151_0:001cae_473_g;src=C;pri=4;
2R ProtHint intron 8011796 8011865 2 - . src=P;mult=1;pri=4;al_score=0.12;
2R ProtHint start 234524 234526 1 + . src=P;mult=1;pri=4;al_score=0.08;
The prediction of all hints with src=M will be enforced. Hints with src=C are 'chained evidence', ie they will only be incorporated if all members of the group (grp=...) can be incorporated in a single transcript. All other hints have src=P in the last column. Supported features in column 3 are intron , start , stop and CDSpart .
If RNA-Seq (and only RNA-Seq) data is provided to BRAKER as a bam-file, and if the genome is softmasked for repeats, BRAKER can automatically train UTR parameters for AUGUSTUS. After successful training of UTR parameters, BRAKER will automatically predict genes including coverage information form RNA-Seq data. Example call:
braker.pl --species=yourSpecies --genome=genome.fasta
--bam=file.bam --UTR=on
Warnings:
This feature is experimental!
--UTR=on is currently not compatible with bamToWig.py as released in AUGUSTUS 3.3.3; it requires the current development code version from the github repository (git clone https://github.com/Gaius-Augustus/Augustus.git).
--UTR=on increases memory consumption of AUGUSTUS. Carefully monitor jobs if your machine was close to maxing RAM without --UTR=on! Reducing the number of cores will also reduce RAM consumption.
UTR prediction sometimes improves coding sequence prediction accuracy, but not always. If you try this feature, carefully compare results with and without UTR parameters, afterwards (eg in UCSC Genome Browser).
For running BRAKER without UTR parameters, it is not very important whether RNA-Seq data was generated by a stranded protocol (because spliced alignments are 'artificially stranded' by checking the splice site pattern). However, for UTR training and prediction, stranded libraries may provide information that is valuable for BRAKER.
After alignment of the stranded RNA-Seq libraries, separate the resulting bam file entries into two files: one for plus strand mappings, one for minus strand mappings. Call BRAKER as follows:
braker.pl --species=yourSpecies --genome=genome.fasta
--bam=plus.bam,minus.bam --stranded=+,-
--UTR=on
You may additionally include bam files from unstranded libraries. Those files will not used for generating UTR training examples, but they will be included in the final gene prediction step as unstranded coverage information, example call:
braker.pl --species=yourSpecies --genome=genome.fasta
--bam=plus.bam,minus.bam,unstranded.bam
--stranded=+,-,. --UTR=on
Warning: This feature is experimental and currently has low priority on our maintenance list!
The native mode for running BRAKER with RNA-Seq and protein data. This will call GeneMark-ETP, which will use RNA-Seq and protein hints for training GeneMark-ETP. Subsequently, AUGUSTUS is trained on 'high-confindent' genes (genes with very high extrinsic evidence support) from the GeneMark-ETP prediction and a set of genes is predicted by AUGUSTUS. In a last step, the predictions of AUGUSTUS and GeneMark-ETP are combined using TSEBRA.
Alignment of RNA-Seq reads
GeneMark-ETP utilizes Stringtie2 to assemble RNA-Seq data, which requires that the aligned reads (BAM files) contain the XS (strand) tag for spliced reads. Therefore, if you align your reads with HISAT2, you must enable the --dta option, or if you use STAR, you must use the --outSAMstrandField intronMotif option. TopHat alignments include this tag by default.
To call the pipeline in this mode, you have to provide it with a protein database using --prot_seq (as described in BRAKER with protein data), and RNA-Seq data either by their SRA ID so that they are downloaded by BRAKER, as unaligned reads in FASTQ format, and/or as aligned reads in bam format (as described in BRAKER with RNA-Seq data). You could also specify already processed extrinsic evidence using the --hints option. However, this is not recommend for a normal BRAKER run in ETPmode, as these hints won't be used in the GeneMark-ETP step. Only use --hints when you want to skip the GenMark-ETP step!
Examples of how you could run BRAKER in ETPmode:
braker.pl --genome=genome.fa --prot_seq=orthodb.fa
--rnaseq_sets_ids=SRA_ID1,SRA_ID2
--rnaseq_sets_dirs=/path/to/local/RNA-Seq/files/
braker.pl --genome=genome.fa --prot_seq=orthodb.fa
--rnaseq_sets_ids=SRA_ID1,SRA_ID2,SRA_ID3
braker.pl --genome=genome.fa --prot_seq=orthodb.fa
--bam=/path/to/SRA_ID1.bam,/path/to/SRA_ID2.bam
A preliminary protocol for integration of assembled subreads from PacBio ccs sequencing in combination with short read Illumina RNA-Seq and protein database is described at https://github.com/Gaius-Augustus/BRAKER/blob/master/docs/long_reads/long_read_protocol .Maryland
We forked GeneMark-ETP and hard coded that StringTie will perform long read assembly in that particular version. If you want to use this 'fast-hack' version for BRAKER, you have to prepare the BAM file with long read to genome spliced alignments outside of BRAKER, eg:
T=48 # adapt to your number of threads
minimap2 -t${T} -ax splice:hq -uf genome.fa isoseq.fa > isoseq.sam
samtools view -bS --threads ${T} isoseq.sam -o isoseq.bam
Pull the adapted container:
singularity build braker3_lr.sif docker://teambraker/braker3:isoseq
Calling BRAKER3 with a BAM file of spliced-aligned IsoSeq Reads:
singularity exec -B ${PWD}:${PWD} braker3_lr.sif braker.pl --genome=genome.fa --prot_seq=protein_db.fa –-bam=isoseq.bam --threads=${T}
Warning Do NOT mix short read and long read data in this BRAKER/GeneMark-ETP variant!
Warning The accuracy of gene prediction here heavily depends on the depth of your isoseq data. We verified with PacBio HiFi reads from 2022 that given sufficient completeness of the assembled transcriptome you will reach similar results as with short reads. However, we also observed a drop in accuracy compared to short reads when using other long read data sets with higher error rates and less sequencing depth.
Please run braker.pl --help to obtain a full list of options.
Compute AUGUSTUS ab initio predictions in addition to AUGUSTUS predictions with hints (additional output files: augustus.ab_initio.* . This may be useful for estimating the quality of training gene parameters when inspecting predictions in a Browser.
One or several command line arguments to be passed to AUGUSTUS, if several arguments are given, separate them by whitespace, ie "--first_arg=sth --second_arg=sth" . This may be be useful if you know that gene prediction in your particular species benefits from a particular AUGUSTUS argument during the prediction step.
Specifies the maximum number of threads that can be used during computation. BRAKER has to run some steps on a single thread, others can take advantage of multiple threads. If you use more than 8 threads, this will not speed up all parallelized steps, in particular, the time consuming optimize_augustus.pl will not use more than 8 threads. However, if you don't mind some threads being idle, using more than 8 threads will speed up other steps.
GeneMark-ETP option: run algorithm with branch point model. Use this option if you genome is a fungus.
Use the present config and parameter files if they exist for 'species'; will overwrite original parameters if BRAKER performs an AUGUSTUS training.
Execute CRF training for AUGUSTUS; resulting parameters are only kept for final predictions if they show higher accuracy than HMM parameters. This increases runtime!
Change the parameter
Generate UTR training examples for AUGUSTUS from RNA-Seq coverage information, train AUGUSTUS UTR parameters and predict genes with AUGUSTUS and UTRs, including coverage information for RNA-Seq as evidence. This is an experimental feature!
If you performed a BRAKER run without --UTR=on, you can add UTR parameter training and gene prediction with UTR parameters (and only RNA-Seq hints) with the following command:
braker.pl --genome=../genome.fa --addUTR=on
--bam=../RNAseq.bam --workingdir=$wd
--AUGUSTUS_hints_preds=augustus.hints.gtf
--threads=8 --skipAllTraining --species=somespecies
Modify augustus.hints.gtf to point to the AUGUSTUS predictions with hints from previous BRAKER run; modify flaning_DNA value to the flanking region from the log file of your previous BRAKER run; modify some_new_working_directory to the location where BRAKER should store results of the additional BRAKER run; modify somespecies to the species name used in your previous BRAKER run.
Add UTRs from RNA-Seq converage information to AUGUSTUS gene predictions using GUSHR. No training of UTR parameters and no gene prediction with UTR parameters is performed.
If you performed a BRAKER run without --addUTR=on, you can add UTRs results of a previous BRAKER run with the following command:
braker.pl --genome=../genome.fa --addUTR=on
--bam=../RNAseq.bam --workingdir=$wd
--AUGUSTUS_hints_preds=augustus.hints.gtf --threads=8
--skipAllTraining --species=somespecies
Modify augustus.hints.gtf to point to the AUGUSTUS predictions with hints from previous BRAKER run; modify some_new_working_directory to the location where BRAKER should store results of the additional BRAKER run; this run will not modify AUGUSTUS parameters. We recommend that you specify the original species of the original run with --species=somespecies . Otherwise, BRAKER will create an unneeded species parameters directory Sp_* .
If --UTR=on is enabled, strand-separated bam-files can be provided with --bam=plus.bam,minus.bam . In that case, --stranded=... should hold the strands of the bam files ( + for plus strand, - for minus strand, . for unstranded). Note that unstranded data will be used in the gene prediction step, only, if the parameter --stranded=... is set. This is an experimental feature! GUSHR currently does not take advantage of stranded data.
If --makehub and [email protected] (with your valid e-mail adress) are provided, a track data hub for visualizing results with the UCSC Genome Browser will be generated using MakeHub (https://github.com/Gaius-Augustus/MakeHub).
By default, GeneMark-ES/ET/EP/ETP uses a probability of 0.001 for predicting the donor splice site pattern GC (instead of GT). It may make sense to increase this value for species where this donor splice site is more common. For example, in the species Emiliania huxleyi , about 50% of donor splice sites have the pattern GC (https://media.nature.com/original/nature-assets/nature/journal/v499/n7457/extref/nature12221-s2.pdf, page 5).
Use a species-specific lineage, eg arthropoda_odb10 for an arthropod. BRAKER does not support auto-typing of the lineage.
Specifying a BUSCO-lineage invokes two changes in BRAKER R28 :
BRAKER will run compleasm with the specified lineage in genome mode and convert the detected BUSCO matches into hints for AUGUSTUS. This may increase the number of BUSCOs in the augustus.hints.gtf file slightly.
BRAKER will invoke best_by_compleasm.py to check whether the braker.gtf file that is by default generated by TSEBRA has the lowest amount of missing BUSCOs compared to the augustus.hints.gtf and the genemark.gtf file. If not, the following decision schema is applied to re-run TSEBRA to minimize the missing BUSCOs in the final output of BRAKER (always braker.gtf). If an alternative and better gene set is created, the original braker.gtf gene set is moved to a directory called braker_original. Information on what happened during the best_by_compleasm.py run is written to the file best_by_compleasm.log.
![best_by_busco[fig14]](https://images.downcodes.com/uploads/20250214/img_67aee79a11fd439.png)
Please note that using BUSCO to assess the quality of a gene set, in particular when comparing BRAKER to other pipelines, does not make sense once you specified a BUSCO lineage. We recommend that you use other measures to assess the quality of your gene set, eg by comparing it to a reference gene set or running OMArk.
BRAKER produces several important output files in the working directory.
braker.gtf: Final gene set of BRAKER. This file may contain different contents depending on how you called BRAKER
in ETPmode: Final gene set of BRAKER consisting of genes predicted by AUGUSTUS and GeneMark-ETP that were combined and filtered by TSEBRA.
otherwise: Union of augustus.hints.gtf and reliable GeneMark-ES/ET/EP predictions (genes fully supported by external evidence). In --esmode , this is the union of augustus.ab_initio.gtf and all GeneMark-ES genes. Thus, this set is generally more sensitive (more genes correctly predicted) and can be less specific (more false-positive predictions can be present). This output is not necessarily better than augustus.hints.gtf, and it is not recommended to use it if BRAKER was run in ESmode.
braker.codingseq: Final gene set with coding sequences in FASTA format
braker.aa: Final gene set with protein sequences in FASTA format
braker.gff3: Final gene set in gff3 format (only produced if the flag --gff3 was specified to BRAKER.
Augustus/*: Augustus gene set(s) in as gtf/conding/aa files
GeneMark-E*/genemark.gtf: Genes predicted by GeneMark-ES/ET/EP/EP+/ETP in GTF-format.
hintsfile.gff: The extrinsic evidence data extracted from RNAseq.bam and/or protein data.
braker_original/*: Genes predicted by BRAKER (TSEBRA merge) before compleasm was used to improve BUSCO completeness
bbc/*: output folder of best_by_compleasm.py script from TSEBRA that is used to improve BUSCO completeness in the final output of BRAKER
Output files may be present with the following name endings and formats:
Coding sequences in FASTA-format are produced if the flag --skipGetAnnoFromFasta was not set.
Protein sequence files in FASTA-format are produced if the flag --skipGetAnnoFromFasta was not set.
For details about gtf format, see http://www.sanger.ac.uk/Software/formats/GFF/. A GTF-format file contains one line per predicted exon. Exemple:
HS04636 AUGUSTUS initial 966 1017 . + 0 transcript_id "g1.1"; gene_id "g1";
HS04636 AUGUSTUS internal 1818 1934 . + 2 transcript_id "g1.1"; gene_id "g1";
The columns (fields) contain:
seqname source feature start end score strand frame transcript ID and gene ID
If the --makehub option was used and MakeHub is available on your system, a hub directory beginning with the name hub_ will be created. Copy this directory to a publicly accessible web server. A file hub.txt resides in the directory. Provide the link to that file to the UCSC Genome Browser for visualizing results.
An incomplete example data set is contained in the directory BRAKER/example . In order to complete the data set, please download the RNA-Seq alignment file (134 MB) with wget :
cd BRAKER/example
wget http://topaz.gatech.edu/GeneMark/Braker/RNAseq.bam
In case you have trouble accessing that file, there's also a copy available from another server:
cd BRAKER/example
wget http://bioinf.uni-greifswald.de/augustus/datasets/RNAseq.bam
The example data set was not compiled in order to achieve optimal prediction accuracy, but in order to quickly test pipeline components. The small subset of the genome used in these test examples is not long enough for BRAKER training to work well.
Data corresponds to the last 1,000,000 nucleotides of Arabidopsis thaliana 's chromosome Chr5, split into 8 artificial contigs.
RNA-Seq alignments were obtained by VARUS.
The protein sequences are a subset of OrthoDB v10 plants proteins.
List of files:
genome.fa - genome file in fasta formatRNAseq.bam - RNA-Seq alignment file in bam format (this file is not a part of this repository, it must be downloaded separately from http://topaz.gatech.edu/GeneMark/Braker/RNAseq.bam)RNAseq.hints - RNA-Seq hints (can be used instead of RNAseq.bam as RNA-Seq input to BRAKER)proteins.fa - protein sequences in fasta formatThe below given commands assume that you configured all paths to tools by exporting bash variables or that you have the necessary tools in your $PATH.
The example data set also contains scripts tests/test*.sh that will execute below listed commands for testing BRAKER with the example data set. You find example results of AUGUSTUS and GeneMark-ES/ET/EP/ETP in the folder results/test* . Be aware that BRAKER contains several parts where random variables are used, ie results that you obtain when running the tests may not be exactly identical. To compare your test results with the reference ones, you can use the compare_intervals_exact.pl script as follows:
# Compare CDS features
compare_intervals_exact.pl --f1 augustus.hints.gtf --f2 ../../results/test${N}/augustus.hints.gtf --verbose
# Compare transcripts
compare_intervals_exact.pl --f1 augustus.hints.gtf --f2 ../../results/test${N}/augustus.hints.gtf --trans --verbose
Several tests use --gm_max_intergenic 10000 option to make the test runs faster. It is not recommended to use this option in real BRAKER runs, the speed increase achieved by adjusting this option is negligible on full-sized genomes.
We give runtime estimations derived from computing on Intel(R) Xeon(R) CPU E5530 @ 2.40GHz .
The following command will run the pipeline according to Figure 3:
braker.pl --genome genome.fa --bam RNAseq.bam --threads N --busco_lineage=lineage_odb10
This test is implemented in test1.sh , expected runtime is ~20 minutes.
The following command will run the pipeline according to Figure 4:
braker.pl --genome genome.fa --prot_seq proteins.fa --threads N --busco_lineage=lineage_odb10
This test is implemented in test2.sh , expected runtime is ~20 minutes.
The following command will run a pipeline that first trains GeneMark-ETP with protein and RNA-Seq hints and subsequently trains AUGUSTUS on the basis of GeneMark-ETP predictions. AUGUSTUS predictions are also performed with hints from both sources, see Figure 5.
Run with local RNA-Seq file:
braker.pl --genome genome.fa --prot_seq proteins.fa --bam ../RNAseq.bam --threads N --busco_lineage=lineage_odb10
This test is implemented in test3.sh , expected runtime is ~20 minutes.
Download RNA-Seq library from Sequence Read Archive (~1gb):
braker.pl --genome genome.fa --prot_seq proteins.fa --rnaseq_sets_ids ERR5767212 --threads N --busco_lineage=lineage_odb10
This test is implemented in test3_4.sh , expected runtime is ~35 minutes.
The training step of all pipelines can be skipped with the option --skipAllTraining . This means, only AUGUSTUS predictions will be performed, using pre-trained, already existing parameters. For example, you can predict genes with the command:
braker.pl --genome=genome.fa --bam RNAseq.bam --species=arabidopsis
--skipAllTraining --threads N
This test is implemented in test4.sh , expected runtime is ~1 minute.
The following command will run the pipeline with no extrinsic evidence:
braker.pl --genome=genome.fa --esmode --threads N
This test is implemented in test5.sh , expected runtime is ~20 minutes.
The following command will run BRAKER with training UTR parameters from RNA-Seq coverage data:
braker.pl --genome genome.fa --bam RNAseq.bam --UTR=on --threads N
This test is implemented in test6.sh , expected runtime is ~20 minutes.
The following command will add UTRs to augustus.hints.gtf from RNA-Seq coverage data:
braker.pl --genome genome.fa --bam RNAseq.bam --addUTR=on --threads N
This test is implemented in test7.sh , expected runtime is ~20 minutes.
There is currently no clean way to restart a failed BRAKER run (after solving some problem). However, it is possible to start a new BRAKER run based on results from a previous run -- given that the old run produced the required intermediate results. We will in the following refer to the old working directory with variable ${BRAKER_OLD} , and to the new BRAKER working directory with ${BRAKER_NEW} . The file what-to-cite.txt will always only refer to the software that was actually called by a particular run. You might have to combine the contents of ${BRAKER_NEW}/what-to-cite.txt with ${BRAKER_OLD}/what-to-cite.txt for preparing a publication. The following figure illustrates at which points BRAKER run may be intercepted.
![braker-intercept[fig8]](https://images.downcodes.com/uploads/20250214/img_67aee79a12cab310.png)
Figure 10: Points for intercepting a BRAKER run and reusing intermediate results in a new BRAKER run.
This option is only possible for BRAKER in ETmode or EPmode and pas in ETPmode!
If you have access to an existing BRAKER output that contains hintsfiles that were generated from extrinsic data, such as RNA-Seq or protein sequences, you can recycle these hints files in a new BRAKER run. Also, hints from a separate ProtHint run can be directly used in BRAKER.
The hints can be given to BRAKER with --hints ${BRAKER_OLD}/hintsfile.gff option. This is illustrated in the test files test1_restart1.sh , test2_restart1.sh , test4_restart1.sh . The other modes (for which this test is missing) cannot be restarted in this way.
The GeneMark result can be given to BRAKER with --geneMarkGtf ${BRAKER_OLD}/GeneMark*/genemark.gtf option if BRAKER is run in ETmode or EPmode. This is illustrated in the test files test1_restart2.sh , test2_restart2.sh , test5_restart2.sh .
In ETPmode, you can either provide BRAKER with the results of the GeneMarkETP step manually, with --geneMarkGtf ${BRAKER_OLD}/GeneMark-ETP/proteins.fa/genemark.gtf , --traingenes ${BRAKER_OLD}/GeneMark-ETP/training.gtf , and --hints ${BRAKER_OLD}/hintsfile.gff (see test3_restart1.sh for an example), or you can specify the previous GeneMark-ETP results with the option --gmetp_results_dir ${BRAKER_OLD}/GeneMark-ETP/ so that BRAKER can search for the files automatically (see test3_restart2.sh for an example).
The trained species parameters for AGUSTUS can be passed with --skipAllTraining and --species $speciesName options. This is illustrated in test*_restart3.sh files. Note that in ETPmode you have to specify the GeneMark files as described in Option 2!
Before reporting bugs, please check that you are using the most recent versions of GeneMark-ES/ET/EP/ETP, AUGUSTUS and BRAKER. Also, check the list of Common problems, and the Issue list on GitHub before reporting bugs. We do monitor open issues on GitHub. Sometimes, we are unable to help you, immediately, but we try hard to solve your problems.
If you found a bug, please open an issue at https://github.com/Gaius-Augustus/BRAKER/issues (or contact [email protected] or [email protected]).
Information worth mentioning in your bug report:
Check in braker/yourSpecies/braker.log at which step braker.pl crashed.
There are a number of other files that might be of interest, depending on where in the pipeline the problem occurred. Some of the following files will not be present if they did not contain any errors.
braker/yourSpecies/errors/bam2hints.*.stderr - will give details on a bam2hints crash (step for converting bam file to intron gff file)
braker/yourSpecies/hintsfile.gff - is this file empty? If yes, something went wrong during hints generation - does this file contain hints from source “b2h” and of type “intron”? If not: GeneMark-ET will not be able to execute properly. Conversely, GeneMark-EP+ will not be able to execute correctly if hints from the source "ProtHint" are missing.
braker/yourSpecies/spaln/*err - errors reported by spaln
braker/yourSpecies/errors/GeneMark-{ET,EP,ETP}.stderr - errors reported by GeneMark-ET/EP+/ETP
braker/yourSpecies/errors/GeneMark-{ET,EP,ETP).stdout - may give clues about the point at which errors in GeneMark-ET/EP+/ETP occured
braker/yourSpecies/GeneMark-{ET,EP,ETP}/genemark.gtf - is this file empty? If yes, something went wrong during executing GeneMark-ET/EP+/ETP
braker/yourSpecies/GeneMark-{ET,EP}/genemark.f.good.gtf - is this file empty? If yes, something went wrong during filtering GeneMark-ET/EP+ genes for training AUGUSTUS
braker/yourSpecies/genbank.good.gb - try a “grep -c LOCUS genbank.good.gb” to determine the number of training genes for training AUGUSTUS, should not be low
braker/yourSpecies/errors/firstetraining.stderr - contains errors from first iteration of training AUGUSTUS
braker/yourSpecies/errors/secondetraining.stderr - contains errors from second iteration of training AUGUSTUS
braker/yourSpecies/errors/optimize_augustus.stderr - contains errors optimize_augustus.pl (additional training set for AUGUSTUS)
braker/yourSpecies/errors/augustus*.stderr - contain AUGUSTUS execution errors
braker/yourSpecies/startAlign.stderr - if you provided a protein fasta file, something went wrong during protein alignment
braker/yourSpecies/startAlign.stdout - may give clues on at which point protein alignment went wrong
BRAKER complains that the RNA-Seq file does not correspond to the provided genome file, but I am sure the files correspond to each other!
Please check the headers of the genome FASTA file. If the headers are long and contain whitespaces, some RNA-Seq alignment tools will truncate sequence names in the BAM file. This leads to an error with BRAKER. Solution: shorten/simplify FASTA headers in the genome file before running the RNA-Seq alignment and BRAKER.
GeneMark fails!
(a) GeneMark by default only uses contigs longer than 50k for training. If you have a highly fragmented assembly, this might lead to "no data" for training. You can override the default minimal length by setting the BRAKER argument --min_contig=10000 .
(b) see "[something] failed to execute" below.
[something] failed to execute!
When providing paths to software to BRAKER, please use absolute, non-abbreviated paths. For example, BRAKER might have problems with --SAMTOOLS_PATH=./samtools/ or --SAMTOOLS_PATH=~/samtools/ . Please use SAMTOOLS_PATH=/full/absolute/path/to/samtools/ , instead. This applies to all path specifications as command line options to braker.pl . Relative paths and absolute paths will not pose problems if you export a bash variable, instead, or if you append the location of tools to your $PATH variable.
GeneMark-ETP in BRAKER dies with '/scratch/11232323': No such file or directory.
This appears to be related to sorting large files, and it's a system configuration depending problem. Solve it with export TMPDIR=/tmp/ before calling BRAKER via Singularity.
BRAKER cannot find the Augustus script XYZ...
Update Augustus from github with git clone https://github.com/Gaius-Augustus/Augustus.git . Do not use Augustus from other sources. BRAKER is highly dependent on an up-to-date Augustus. Augustus releases happen rather rarely, updates to the Augustus scripts folder occur rather frequently.
Does BRAKER depend on Python3?
It does. The python scripts employed by BRAKER are not compatible with Python2.
Why does BRAKER predict more genes than I expected?
If transposable elements (or similar) have not been masked appropriately, AUGUSTUS tends to predict those elements as protein coding genes. This can lead to a huge number genes. You can check whether this is the case for your project by BLASTing (or DIAMONDing) the predicted protein sequences against themselves (all vs. all) and counting how many of the proteins have a high number of high quality matches. You can use the output of this analysis to divide your gene set into two groups: the protein coding genes that you want to find and the repetitive elements that were additionally predicted.
I am running BRAKER in Anaconda and something fails...
Update AUGUSTUS and BRAKER from github with git clone https://github.com/Gaius-Augustus/Augustus.git and git clone https://github.com/Gaius-Augustus/BRAKER.git . The Anaconda installation is great, but it relies on releases of AUGUSTUS and BRAKER - which are often lagging behind. Please use the current GitHub code, instead.
Why and where is the GenomeThreader support gone?
BRAKER is a joint project between teams from University of Greifswald and Georgia Tech. While the group of Mark Bordovsky from Georgia Tech contributes GeneMark expertise, the group of Mario Stanke from University of Greifswald contributes AUGUSTUS expertise. Using GenomeThreader to build training genes for AUGUSTUS in BRAKER circumvents execution of GeneMark. Thus, the GenomeThreader mode is strictly speaking not part of the BRAKER project. The previous functionality of BRAKER with GenomeThreader has been moved to GALBA at https://github.com/Gaius-Augustus/GALBA. Note that GALBA has also undergone extension for using Miniprot instead of GenomeThreader.
My BRAKER gene set has too many BUSCO duplicates!
AUGUSTUS within BRAKER can predict alternative splicing isoforms. Also the merge of the AUGUSTUS and GeneMark gene set by TSEBRA within BRAKER may result in additional isoforms for a single gene. The BUSCO duplicates usually come from alternative splicing isoforms, ie they are expected.
Augustus and/or etraining within BRAKER complain that the file aug_cmdln_parameters.json is missing. Even though I am using the latest Singularity container!
BRAKER copies the AUGUSTUS_CONFIG_PATH folder to a writable location. In older versions of Augustus, that file was indeed not existing. If the local writable copy of a folder already exists, BRAKER will not re-copy it. Simply delete the old folder. (It is often ~/.augustus , so you can simply do rm -rf ~/.augustus ; the folder might be residing in $PWD if your home directory was not writable).
I sit behind a firewall, compleasm cannot download the BUSCO files, what can I do? See Issue #785 (comment)
Since BRAKER is a pipeline that calls several Bioinformatics tools, publication of results obtained by BRAKER requires that not only BRAKER is cited, but also the tools that are called by BRAKER. BRAKER will output a file what-to-cite.txt in the BRAKER working directory, informing you about which exact sources apply to your run.
Always cite:
Stanke, M., Diekhans, M., Baertsch, R. and Haussler, D. (2008). Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics, doi: 10.1093/bioinformatics/btn013.
Stanke. M., Schöffmann, O., Morgenstern, B. and Waack, S. (2006). Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics 7, 62.
If you provided any kind of evidence for BRAKER, cite:
If you provided both short read RNA-Seq evidence and a large database of proteins, cite:
Gabriel, L., Bruna, T., Hoff, KJ, Ebel, M., Lomsadze, A., Borodovsky, M., Stanke, M. (2023). BRAKER3: Fully Automated Genome Annotation Using RNA-Seq and Protein Evidence with GeneMark-ETP, AUGUSTUS and TSEBRA. bioRxiV, doi: 10.1101/2023.06.10.54444910.1101/2023.01.01.474747.
Bruna, T., Lomsadze, A., Borodovsky, M. (2023). GeneMark-ETP: Automatic Gene Finding in Eukaryotic Genomes in Consistence with Extrinsic Data. bioRxiv, doi: 10.1101/2023.01.13.524024.
Kovaka, S., Zimin, AV, Pertea, GM, Razaghi, R., Salzberg, SL, & Pertea, M. (2019). Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome biology, 20(1):1-13.
Pertea, G., & Pertea, M. (2020). GFF utilities: GffRead and GffCompare. F1000Research, 9.
Quinlan, AR (2014). BEDTools: the Swiss‐army tool for genome feature analysis. Current protocols in bioinformatics, 47(1):11-12.
If the only source of evidence for BRAKER was a large database of protein sequences, cite:
If the only source of evidence for BRAKER was RNA-Seq data, cite:
Hoff, KJ, Lange, S., Lomsadze, A., Borodovsky, M. and Stanke, M. (2016). BRAKER1: unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics, 32(5):767-769.
Lomsadze, A., Paul DB, and Mark B. (2014) Integration of Mapped Rna-Seq Reads into Automatic Training of Eukaryotic Gene Finding Algorithm. Nucleic Acids Research 42(15): e119--e119
If you called BRAKER3 with an IsoSeq BAM file, or if you envoked the --busco_lineage option, cite:
If you called BRAKER with the --busco_lineage option, in addition, cite:
Simão, FA, Waterhouse, RM, Ioannidis, P., Kriventseva, EV, & Zdobnov, EM (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics, 31(19), 3210-3212.
Li, H. (2023). Protein-to-genome alignment with miniprot. Bioinformatics, 39(1), btad014.
Huang, N., & Li, H. (2023). compleasm: a faster and more accurate reimplementation of BUSCO. Bioinformatics, 39(10), btad595.
If any kind of AUGUSTUS training was performed by BRAKER, check carefully whether you configured BRAKER to use NCBI BLAST or DIAMOND. One of them was used to filter out redundant training gene structures.
If you used NCBI BLAST, please cite:
Altschul, AF, Gish, W., Miller, W., Myers, EW and Lipman, DJ (1990). A basic local alignment search tool. J Mol Biol 215:403--410.
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., and Madden, TL (2009). Blast+: architecture and applications. BMC bioinformatics, 10(1):421.
If you used DIAMOND, please cite:
If BRAKER was executed with a genome file and no extrinsic evidence, cite, then GeneMark-ES was used, cite:
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, YO and Borodovsky, M. (2005). Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Research, 33(20):6494--6506.
Ter-Hovhannisyan, V., Lomsadze, A., Chernoff, YO and Borodovsky, M. (2008). Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome research, pages gr--081612, 2008.
Hoff, KJ, Lomsadze, A., Borodovsky, M. and Stanke, M. (2019). Whole-Genome Annotation with BRAKER. Methods Mol Biol. 1962:65-95, doi: 10.1007/978-1-4939-9173-0_5.
If BRAKER was run with proteins as source of evidence, please cite all tools that are used by the ProtHint pipeline to generate hints:
Bruna, T., Lomsadze, A., & Borodovsky, M. (2020). GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genomics and Bioinformatics, 2(2), lqaa026.
Buchfink, B., Xie, C., Huson, DH (2015). Fast and sensitive protein alignment using DIAMOND. Nature Methods 12:59-60.
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, YO and Borodovsky, M. (2005). Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Research, 33(20):6494--6506.
Iwata, H., and Gotoh, O. (2012). Benchmarking spliced alignment programs including Spaln2, an extended version of Spaln that incorporates additional species-specific features. Nucleic acids research, 40(20), e161-e161.
Gotoh, O., Morita, M., Nelson, DR (2014). Assessment and refinement of eukaryotic gene structure prediction with gene-structure-aware multiple protein sequence alignment. BMC bioinformatics, 15(1), 189.
If BRAKER was executed with RNA-Seq alignments in bam-format, then SAMtools was used, cite:
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R.; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25(16):2078-9.
Barnett, DW, Garrison, EK, Quinlan, AR, Strömberg, MP and Marth GT (2011). BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics, 27(12):1691-2
If BRAKER downloaded RNA-Seq libraries from SRA using their IDs, cite SRA, SRA toolkit, and HISAT2:
Leinonen, R., Sugawara, H., Shumway, M., & International Nucleotide Sequence Database Collaboration. (2010). The sequence read archive. Nucleic acids research, 39(suppl_1), D19-D21.
SRA Toolkit Development Team (2020). SRA Toolkit. https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software.
Kim, D., Paggi, JM, Park, C., Bennett, C., & Salzberg, SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature biotechnology, 37(8):907-915.
If BRAKER was executed using RNA-Seq data in FASTQ format, cite HISAT2:
If BRAKER called MakeHub for creating a track data hub for visualization of BRAKER results with the UCSC Genome Browser, cite:
If BRAKER called GUSHR for generating UTRs, cite:
Keilwagen, J., Hartung, F., Grau, J. (2019) GeMoMa: Homology-based gene prediction utilizing intron position conservation and RNA-seq data. Methods Mol Biol. 1962:161-177, doi: 10.1007/978-1-4939-9173-0_9.
Keilwagen, J., Wenk, M., Erickson, JL, Schattat, MH, Grau, J., Hartung F. (2016) Using intron position conservation for homology-based gene prediction. Nucleic Acids Research, 44(9):e89.
Keilwagen, J., Hartung, F., Paulini, M., Twardziok, SO, Grau, J. (2018) Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinformatics, 19(1):189.
All source code, ie scripts/*.pl or scripts/*.py are under the Artistic License (see http://www.opensource.org/licenses/artistic-license.php).
[F1] EX = ES/ET/EP/ETP, all available for download under the name GeneMark-ES/ET/EP ↩
[F2] Please use the latest version from the master branch of AUGUSTUS distributed by the original developers, it is available from github at https://github.com/Gaius-Augustus/Augustus. Problems have been reported from users that tried to run BRAKER with AUGUSTUS releases maintained by third parties, ie Bioconda. ↩
[F4] install with sudo apt-get install cpanminus ↩
[F6] The binary may eg reside in bamtools/build/src/toolkit ↩
[R0] Bruna, Tomas, Hoff, Katharina J., Lomsadze, Alexandre, Stanke, Mario, and Borodovsky, Mark. 2021. “BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database." NAR Genomics and Bioinformatics 3(1):lqaa108.↩
[R1] Hoff, Katharina J, Simone Lange, Alexandre Lomsadze, Mark Borodovsky, and Mario Stanke. 2015. “BRAKER1: Unsupervised Rna-Seq-Based Genome Annotation with Genemark-et and Augustus.” Bioinformatics 32 (5). Oxford University Press: 767--69.↩
[R2] Lomsadze, Alexandre, Paul D Burns, and Mark Borodovsky. 2014. “Integration of Mapped Rna-Seq Reads into Automatic Training of Eukaryotic Gene Finding Algorithm.” Nucleic Acids Research 42 (15). Oxford University Press: e119--e119.↩
[R3] Stanke, Mario, Mark Diekhans, Robert Baertsch, and David Haussler. 2008. “Using Native and Syntenically Mapped cDNA Alignments to Improve de Novo Gene Finding.” Bioinformatics 24 (5). Oxford University Press: 637--44.↩
[R4] Stanke, Mario, Oliver Schöffmann, Burkhard Morgenstern, and Stephan Waack. 2006. “Gene Prediction in Eukaryotes with a Generalized Hidden Markov Model That Uses Hints from External Sources.” BMC Bioinformatics 7 (1). BioMed Central: 62.↩
[R5] Barnett, Derek W, Erik K Garrison, Aaron R Quinlan, Michael P Strömberg, and Gabor T Marth. 2011. “BamTools: A C++ Api and Toolkit for Analyzing and Managing Bam Files.” Bioinformatics 27 (12). Oxford University Press: 1691--2.↩
[R6] Li, Heng, Handsaker, Bob, Alec Wysoker, Tim Fennell, Jue Ruan, Nils Homer, Gabor Marth, Goncalo Abecasis, and Richard Durbin. 2009. “The Sequence Alignment/Map Format and Samtools.” Bioinformatics 25 (16). Oxford University Press: 2078--9.↩
[R7] Gremme, G. 2013. “Computational Gene Structure Prediction.” PhD thesis, Universität Hamburg.↩
[R8] Gotoh, Osamu. 2008a. “A Space-Efficient and Accurate Method for Mapping and Aligning cDNA Sequences onto Genomic Sequence.” Nucleic Acids Research 36 (8). Oxford University Press: 2630--8.↩
[R9] Iwata, Hiroaki, and Osamu Gotoh. 2012. “Benchmarking Spliced Alignment Programs Including Spaln2, an Extended Version of Spaln That Incorporates Additional Species-Specific Features.” Nucleic Acids Research 40 (20). Oxford University Press: e161--e161.↩
[R10] Osamu Gotoh. 2008b. “Direct Mapping and Alignment of Protein Sequences onto Genomic Sequence.” Bioinformatics 24 (21). Oxford University Press: 2438--44.↩
[R11] Slater, Guy St C, and Ewan Birney. 2005. “Automated Generation of Heuristics for Biological Sequence Comparison.” BMC Bioinformatics 6(1). BioMed Central: 31.↩
[R12] Altschul, SF, W. Gish, W. Miller, EW Myers, and DJ Lipman. 1990. “Basic Local Alignment Search Tool.” Journal of Molecular Biology 215:403--10.↩
[R13] Camacho, Christiam, et al. 2009. “BLAST+: architecture and applications.“ BMC Bioinformatics 1(1): 421.↩
[R14] Lomsadze, A., V. Ter-Hovhannisyan, YO Chernoff, and M. Borodovsky. 2005. “Gene identification in novel eukaryotic genomes by self-training algorithm.” Nucleic Acids Research 33 (20): 6494--6506. doi:10.1093/nar/gki937.↩
[R15] Ter-Hovhannisyan, Vardges, Alexandre Lomsadze, Yury O Chernoff, and Mark Borodovsky. 2008. “Gene Prediction in Novel Fungal Genomes Using an Ab Initio Algorithm with Unsupervised Training.” Genome Research . Cold Spring Harbor Lab, gr--081612.↩
[R16] Hoff, KJ 2019. MakeHub: Fully automated generation of UCSC Genome Browser Assembly Hubs. Genomics, Proteomics and Bioinformatics , in press, preprint on bioarXive, doi: https://doi.org/10.1101/550145.↩
[R17] Bruna, T., Lomsadze, A., & Borodovsky, M. 2020. GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genomics and Bioinformatics, 2(2), lqaa026. doi: https://doi.org/10.1093/nargab/lqaa026.↩
[R18] Kriventseva, EV, Kuznetsov, D., Tegenfeldt, F., Manni, M., Dias, R., Simão, FA, and Zdobnov, EM 2019. OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Research, 47(D1), D807-D811.↩
[R19] Keilwagen, J., Hartung, F., Grau, J. (2019) GeMoMa: Homology-based gene prediction utilizing intron position conservation and RNA-seq data. Methods Mol Biol. 1962:161-177, doi: 10.1007/978-1-4939-9173-0_9.↩
[R20] Keilwagen, J., Wenk, M., Erickson, JL, Schattat, MH, Grau, J., Hartung F. (2016) Using intron position conservation for homology-based gene prediction. Nucleic Acids Research, 44(9):e89.↩
[R21] Keilwagen, J., Hartung, F., Paulini, M., Twardziok, SO, Grau, J. (2018) Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinformatics, 19(1):189.↩
[R22] SRA Toolkit Development Team (2020). SRA Toolkit. https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software.[↩](#a22)
[R23] Kim, D., Paggi, JM, Park, C., Bennett, C., & Salzberg, SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature biotechnology, 37(8):907-915.↩
[R24] Quinlan, AR (2014). BEDTools: the Swiss‐army tool for genome feature analysis. Current protocols in bioinformatics, 47(1):11-12.↩
[R25] Kovaka, S., Zimin, AV, Pertea, GM, Razaghi, R., Salzberg, SL, & Pertea, M. (2019). Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome biology, 20(1):1-13.↩
[R26] Pertea, G., & Pertea, M. (2020). GFF utilities: GffRead and GffCompare. F1000Research, 9.↩
[R27] Huang, N., & Li, H. (2023). compleasm: a faster and more accurate reimplementation of BUSCO. Bioinformatics, 39(10), btad595.↩
[R28] Bruna, T., Gabriel, L. & Hoff, KJ (2024). Navigating Eukaryotic Genome Annotation Pipelines: A Route Map to BRAKER, Galba, and TSEBRA. arXiv, https://doi.org/10.48550/arXiv.2403.19416 .↩


