reference_database_creator
bug fix --in-silico-pcr --untrimmed
KEPITING ( C makan R database referensi untuk A mplikon- B ased S quencing) adalah program perangkat lunak serbaguna yang menghasilkan database referensi yang dikurasi untuk analisis metagenomik. Alur kerja CRABS terdiri dari tujuh modul: (i) mengunduh data dari repositori online; (ii) mengimpor data yang diunduh ke dalam format CRABS; (iii) mengekstrak daerah amplikon melalui analisis in silico PCR; (iv) mengambil amplikon tanpa daerah pengikatan primer melalui penyelarasan dengan kode batang yang diekstraksi secara in silico ; (v) menyusun dan mengelompokkan basis data lokal melalui beberapa parameter pemfilteran; (vi) mengekspor database lokal dalam berbagai format sesuai dengan persyaratan pengklasifikasi taksonomi; dan (vi) fungsi pasca-pemrosesan, yaitu visualisasi, untuk mengeksplorasi dan memberikan gambaran ringkasan database referensi lokal. Ketujuh modul ini dibagi menjadi delapan belas fungsi dan dijelaskan di bawah ini. Selain itu, kode contoh disediakan untuk masing-masing dari delapan belas fungsi. Terakhir, tutorial untuk membangun database referensi hiu lokal untuk set primer MiFish-E disediakan di akhir dokumen README ini untuk memberikan contoh skrip sebagai referensi.
Kami sangat gembira mengumumkan bahwa CRABS telah melihat pembaruan besar dan desain ulang kode berdasarkan umpan balik pengguna, yang kami harap akan meningkatkan pengalaman pengguna dalam membangun basis data referensi lokal Anda sendiri!
Silakan temukan di bawah ini daftar fitur dan peningkatan yang ditambahkan ke CRABS v 1.0.0 :
CRABS v 1.0.0 sekarang dapat diunduh secara manual dengan mengkloning repositori GitHub ini (lihat 4.1 Instalasi manual untuk info detail). Kami akan memperbarui container Docker dan paket conda sesegera mungkin untuk memudahkan instalasi versi terbaru.
Saat menggunakan CRABS dalam proyek penelitian Anda, harap mengutip makalah berikut:
[Jeunen, G.-J., Dowle, E., Edgecombe, J., von Ammon, U., Gemmell, N. J., & Cross, H. (2022). crabs—A software program to generate curated reference databases for metabarcoding sequencing data. Molecular Ecology Resources, 00, 1– 14.](https://doi.org/10.1111/1755-0998.13741)
CRABS adalah toolkit baris perintah yang berjalan pada lingkungan Unix/Linux biasa dan secara eksklusif ditulis dalam python3. Namun, CRABS memanfaatkan modul subproses di python untuk menjalankan beberapa perintah dalam sintaks bash untuk menghindari keanehan khusus python dan meningkatkan kecepatan eksekusi. Kami menyediakan tiga cara untuk menginstal CRABS. Untuk versi CRABS terbaru, kami merekomendasikan instalasi manual dengan mengkloning repositori GitHub ini dan menginstal 10 dependensi secara terpisah (instruksi instalasi untuk semua dependensi disediakan di 4.1 Instalasi manual). CRABS juga dapat diinstal melalui Docker dan conda. Kedua metode ini memungkinkan instalasi yang mudah dengan menginstal semua dependensi secara otomatis. Kami bertujuan untuk selalu memperbarui kontainer Docker dan paket conda, meskipun penundaan tertentu untuk memperbarui ke versi terbaru dapat terjadi, terutama untuk paket conda. Di bawah ini adalah rincian untuk ketiga pendekatan tersebut.
Untuk instalasi manual, clone terlebih dahulu repositori CRABS. Langkah ini mengharuskan GitHub tersedia di baris perintah (petunjuk instalasi untuk GitHub).
git clone https://github.com/gjeunen/reference_database_creator.git
Tergantung pada pengaturan Anda, CRABS mungkin perlu dibuat dapat dieksekusi di sistem Anda. Hal ini dapat dicapai dengan menggunakan kode di bawah ini.
chmod +x reference_database_creator/crabs
Setelah CRABS terinstal, kita perlu memastikan semua dependensi terinstal dan dapat diakses secara global. Versi terbaru CRABS (versi v 1.0.0 ) berjalan pada Python 3.11.7 (atau versi apa pun yang kompatibel dengan 3.11.7) dan bergantung pada lima modul Python yang mungkin tidak standar dengan Python, serta lima program perangkat lunak eksternal. Semua dependensi tercantum di bawah, bersama dengan tautan ke petunjuk instalasi. Nomor versi yang diberikan untuk setiap modul dan program perangkat lunak adalah nomor versi yang dikembangkan oleh CRABS. Meskipun versi yang kompatibel dari masing-masing versi dapat digunakan juga.
Modul Python:
Program perangkat lunak eksternal:
Setelah CRABS dan semua dependensinya diinstal, CRABS dapat diakses di seluruh OS menggunakan kode di bawah ini.
export PATH="/path/to/crabs/folder:$PATH"
Gantikan /path/to/crabs/folder dengan jalur sebenarnya ke folder repositori GitHub di OS, yaitu folder yang dibuat selama perintah git clone di atas. Menambahkan kode export ke file .bash_profile atau .bashrc akan membuat CRABS dapat diakses secara global kapan saja.
Docker adalah proyek sumber terbuka yang memungkinkan penerapan aplikasi perangkat lunak di dalam 'wadah' yang diisolasi dari komputer Anda dan dijalankan melalui sistem operasi host virtual yang disebut Docker Engine. Keuntungan utama menjalankan buruh pelabuhan dibandingkan mesin virtual adalah mereka menggunakan sumber daya yang jauh lebih sedikit. Isolasi ini berarti Anda dapat menjalankan container Docker di sebagian besar sistem operasi, termasuk Mac, Windows, dan Linux. Anda mungkin perlu menyiapkan akun gratis untuk menggunakan Docker Desktop. Tautan ini memiliki pengenalan yang bagus tentang dasar-dasar penggunaan Docker. Berikut ini tautan untuk membantu Anda memulai dan berorientasi pada multiverse Docker.
Hanya ada dua langkah untuk menjalankan Crabs di komputer Anda. Pertama, instal Docker Desktop di komputer Anda, yang gratis untuk sebagian besar pengguna. Berikut adalah instruksi untuk Mac ; berikut adalah petunjuk untuk komputer Windows , dan berikut adalah petunjuk untuk Linux (sebagian besar platform Linux utama didukung). Setelah Anda menginstal dan menjalankan Docker Desktop (aplikasi Desktop harus berjalan agar Anda dapat menggunakan perintah buruh pelabuhan apa pun di baris perintah), Anda hanya perlu 'menarik' image Crabs kami, dan Anda siap untuk memulai:
docker pull quay.io/swordfish/crabs:0.1.7
Meskipun instalasi aplikasi buruh pelabuhan mudah, penggunaan aplikasi tersebut mungkin sedikit rumit pada awalnya. Untuk membantu Anda memulai, kami telah menyediakan beberapa contoh perintah menggunakan kepiting versi buruh pelabuhan. Contoh-contoh ini dapat ditemukan di folder docker_intro di repo ini . Dari contoh-contoh ini Anda seharusnya dapat menjalankan pengaturan seluruh database referensi dan siap untuk memulai. Kami akan terus memperluas contoh-contoh ini dan mengujinya dalam berbagai situasi berbeda. Silakan ajukan pertanyaan dan berikan masukan di tab Masalah.
Untuk menginstal paket conda, Anda harus menginstal conda terlebih dahulu. Lihat tautan ini untuk detailnya. Jika conda sudah terinstal, sebaiknya perbarui alat conda dengan conda update conda sebelum menginstal CRABS.
Setelah conda diinstal, ikuti langkah-langkah di bawah ini untuk menginstal CRABS dan semua dependensi. Pastikan untuk memasukkan perintah sesuai urutan kemunculannya di bawah.
conda create -n CRABS
conda activate CRABS
conda config --add channels bioconda
conda config --add channels conda-forge
conda install -c bioconda crabs
Setelah Anda memasukkan perintah install, conda akan memproses permintaan tersebut (ini mungkin memakan waktu sekitar satu menit), dan kemudian menampilkan semua paket dan program yang akan diinstal, dan meminta konfirmasi. Ketik y untuk memulai instalasi. Setelah ini selesai, CRABS harus siap berangkat.
Kami telah menguji instalasi ini pada sistem Mac dan Linux. Kami belum menguji Subsistem Windows untuk Linux (WSL).
Gunakan kode di bawah ini untuk memeriksa apakah CRABS berhasil diinstal dan dapatkan informasi bantuan.
crabs -hInformasi bantuan membagi delapan belas fungsi ke dalam grup berbeda, dengan masing-masing grup mencantumkan fungsi di bagian atas dan parameter wajib dan opsional di bawahnya.

CRABS berisi tujuh modul, yang menggabungkan delapan belas fungsi:
Modul 1: mengunduh data dari repositori online
--download-taxonomy : unduh informasi taksonomi NCBI;--download-bold : mengunduh data urutan dari Barcode of Life Database (BOLD);--download-embl : mengunduh data urutan dari Arsip Nukleotida Eropa (ENA; EMBL);--download-mitofish : mengunduh data urutan dari database MitoFish;--download-ncbi : mengunduh data urutan dari Pusat Informasi Bioteknologi Nasional (NCBI).Modul 2: mengimpor data yang diunduh ke format CRABS
--import : mengimpor urutan unduhan atau kode batang khusus ke dalam format CRABS;--merge : menggabungkan berbagai file berformat CRABS menjadi satu file.Modul 3: mengekstrak daerah amplikon melalui analisis in silico PCR
--in-silico-pcr : mengekstrak amplikon dari data yang diunduh dengan mencari dan menghilangkan daerah pengikatan primer.Modul 4: mengambil amplikon tanpa daerah pengikatan primer
--pairwise-global-alignment : mengambil amplikon tanpa daerah pengikatan primer dengan menyelaraskan urutan yang diunduh ke kode batang yang diekstraksi secara silico .Modul 5: menyusun dan mengelompokkan database lokal melalui beberapa parameter pemfilteran
--dereplicate : membuang urutan duplikat;--filter : membuang urutan melalui beberapa parameter pemfilteran;--subset : subset database lokal untuk mempertahankan atau mengecualikan kelompok taksonomi tertentu.Modul 6: mengekspor database lokal
--export : mengekspor database berformat CRABS ke berbagai format sesuai dengan kebutuhan pengklasifikasi taksonomi yang akan digunakan.Modul 7: fungsi pasca-pemrosesan untuk mengeksplorasi dan memberikan gambaran ringkasan database referensi lokal
--diversity-figure : membuat diagram batang horizontal yang menampilkan jumlah spesies dan kelompok sekuens per level tertentu yang termasuk dalam database referensi;--amplicon-length-figure : membuat diagram garis yang menggambarkan distribusi panjang amplikon yang dipisahkan berdasarkan kelompok taksonomi;--phylogenetic-tree : membuat pohon filogenetik dengan kode batang dari database referensi untuk daftar spesies target;--amplification-efficiency-figure : membuat grafik batang yang menampilkan ketidakcocokan di wilayah pengikatan primer;--completeness-table : membuat spreadsheet yang berisi ketersediaan barcode untuk kelompok taksonomi.Data pengurutan awal dapat diunduh oleh CRABS dari empat repositori online, termasuk (i) BOLD, (ii) EMBL, (iii) MitoFish, dan NCBI. Mulai versi v 1.0.0 dan seterusnya, pengunduhan data dari setiap repositori dibagi menjadi fungsinya masing-masing. Selain itu, CRABS tidak secara otomatis memformat data setelah pengunduhan untuk meningkatkan fleksibilitas dan mengaktifkan debugging ketika pengunduhan data gagal.
Selain mengunduh data sekuens, CRABS juga mampu mengunduh informasi taksonomi NCBI, yang digunakan CRABS untuk membuat garis keturunan taksonomi untuk setiap sekuens.
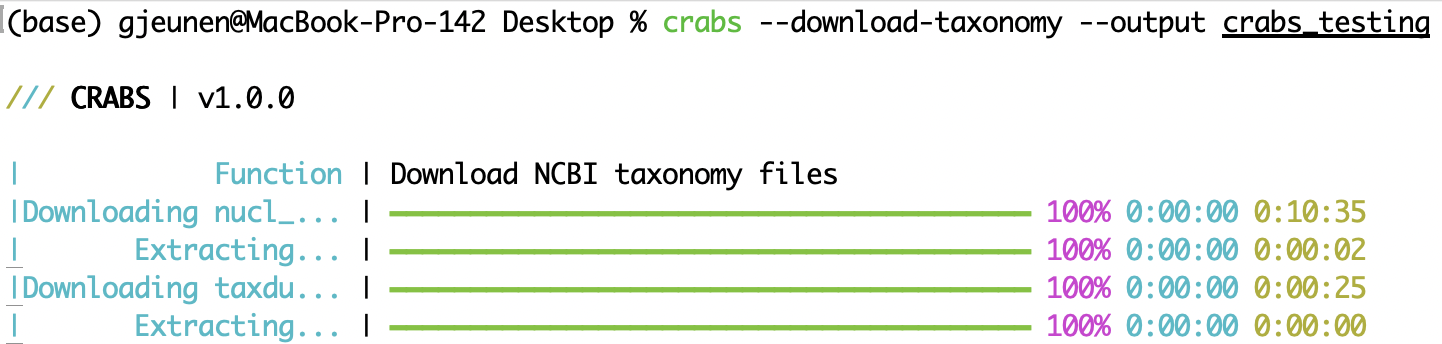
--download-taxonomy Untuk menetapkan garis keturunan taksonomi pada setiap urutan yang diunduh dalam database referensi (lihat 5.2 Modul 2), informasi taksonomi perlu diunduh. CRABS menggunakan taksonomi NCBI dan mengunduh tiga file spesifik ke komputer Anda: (i) file yang menghubungkan nomor aksesi ke ID taksonomi ( nucl_gb.accession2taxid ), (ii) file yang berisi informasi tentang nama filogenetik yang terkait dengan setiap ID taksonomi ( names.dmp ), dan (iii) file yang berisi informasi bagaimana ID taksonomi ditautkan ( node.dmp ). Direktori keluaran untuk file yang diunduh dapat ditentukan menggunakan parameter --output . Untuk mengecualikan file nucl_gb.accession2taxid atau nama file.dmp dan node.dmp , parameter --exclude acc2tax atau --exclude taxdump dapat disediakan. Kode pertama di bawah ini tidak mengunduh file apa pun, karena acc2tax dan taxdump disediakan untuk parameter --exclude . Baris kode kedua mengunduh ketiga file ke subdirektori --output crabs_testing . Tangkapan layar di bawah menampilkan apa yang dicetak ke konsol saat menjalankan baris kode ini.
crabs --download-taxonomy --exclude 'acc2taxid,taxdump'
crabs --download-taxonomy --output crabs_testing
--download-bold Urutan BOLD diunduh melalui situs BOLD. File keluaran, yang disusun sebagai dokumen fasta dua baris, dapat ditentukan menggunakan parameter --output . Pengguna dapat menentukan grup taksonomi mana yang akan diunduh menggunakan parameter --taxon . Kami merekomendasikan penulisan perulangan for sederhana (contoh diberikan di bawah) ketika pengguna ingin mengunduh beberapa grup taksonomi, sehingga membatasi jumlah data yang akan diunduh dari BOLD per instans. Namun, jika hanya sejumlah kecil kelompok taksonomi yang diminati, nama kelompok taksonomi juga dapat dipisahkan dengan | (contoh disediakan di bawah). Kami juga menyarankan pengguna untuk memeriksa apakah nama grup taksonomi yang akan diunduh tercantum dalam arsip BOLD atau apakah nama alternatif perlu digunakan. Misalnya, menentukan --taxon Chondrichthyes tidak akan mengunduh semua rangkaian ikan bertulang rawan dari BOLD, karena nama kelas ini tidak tercantum di BOLD. Pengguna sebaiknya menggunakan --taxon Elasmobranchii dalam hal ini. Pengguna juga dapat menentukan untuk membatasi pengunduhan ke penanda genetik tertentu dengan menyediakan parameter --marker . Jika terdapat beberapa penanda genetik yang diinginkan, nama penanda harus dipisahkan dengan | . Empat penanda barcode DNA utama pada BOLD adalah COI-5P , ITS , matK , dan rbcL . Masukan untuk parameter --marker peka huruf besar-kecil.
Pendekatan yang disarankan: Perulangan sederhana untuk mengunduh data dari BOLD untuk beberapa kelompok taksonomi (pendekatan yang disarankan). Kode di bawah ini pertama-tama mengunduh data untuk Elasmobranchii, diikuti dengan urutan yang ditetapkan ke Mamalia. Data yang diunduh akan ditulis ke subdirektori --output crabs_testing dan ditempatkan dalam dua file terpisah, yang menunjukkan data mana yang termasuk dalam kelompok taksonomi mana, yaitu crabs_testing/bold_Elasmobranchii.fasta dan crabs_testing/bold_Mammalia.fasta .
for taxon in Elasmobranchii Mammalia; do crabs --download-bold --taxon ${taxon} --output crabs_testing/bold_${taxon}.fasta; done

Opsi alternatif: Selain perulangan for yang direkomendasikan, beberapa nama takson dapat diberikan sekaligus dengan memisahkan nama menggunakan | .
crabs --download-bold --taxon 'Elasmobranchii|Mammalia' --output crabs_testing/bold_elasmobranchii_mammalia.fasta
--download-embl Urutan dari EMBL diunduh melalui situs ENA FTP. File EMBL pertama-tama akan diunduh dalam format '.fasta.gz' dan secara otomatis dibuka zipnya setelah unduhan selesai. Basis data ini tidak memberikan banyak fleksibilitas sehubungan dengan pengunduhan selektif dibandingkan dengan BOLD atau NCBI. Sebaliknya, data EMBL disusun menjadi 15 divisi pajak, yang dapat diunduh secara terpisah. Divisi pajak yang akan diunduh dapat ditentukan menggunakan parameter --taxon . Karena setiap divisi pajak dibagi menjadi beberapa file, tanda * diberikan setelah nama untuk mengunduh semua file. Pengguna juga dapat mendownload file tertentu dengan menuliskan nama file secara lengkap. Daftar 15 opsi divisi pajak disediakan di bawah ini. Direktori keluaran dan nama file dapat ditentukan menggunakan parameter --output .
Daftar divisi perpajakan:
crabs --download-embl --taxon 'mam*' --output crabs_testing/embl_mam.fasta
--download-mitofish CRABS juga dapat mengunduh database MitoFish. Basis data ini adalah file fasta dua baris tunggal. Direktori keluaran dan nama file dapat ditentukan menggunakan parameter --output .
crabs --download-mitofish --output crabs_testing/mitofish.fasta

--download-ncbi Urutan dari database NCBI diunduh melalui Entrez Programming Utilities. NCBI memungkinkan pengunduhan data dari berbagai database, yang dapat ditentukan pengguna dengan parameter --database . Bagi sebagian besar pengguna, database --database nucleotide akan menjadi yang paling tepat untuk membangun database referensi lokal.
Untuk menentukan data yang akan diunduh dari NCBI, pengguna menyediakan pencarian melalui parameter --query . Membuat penelusuran NCBI yang baik bisa jadi sulit. Cara yang baik untuk membuat permintaan pencarian adalah dengan menggunakan jendela pencarian halaman web NCBI. Dari tautan ini, lakukan pencarian awal terlebih dahulu dan tekan enter. Ini akan membawa Anda ke halaman hasil di mana Anda dapat mempersempit pencarian Anda lebih lanjut. Pada tangkapan layar di bawah, kami telah menyempurnakan pencarian lebih lanjut dengan membatasi panjang urutan antara 100 - 25.000 bp dan hanya memasukkan urutan mitokondria. Pengguna dapat menyalin-menempelkan teks di kotak "Detail pencarian" di situs web dan memberikannya dalam tanda kutip pada parameter --query . Manfaat lain menggunakan jendela pencarian halaman web NCBI adalah halaman web akan menampilkan berapa banyak urutan yang cocok dengan permintaan pencarian Anda, yang harus sesuai dengan jumlah urutan yang dilaporkan oleh CRABS. Halaman web ini memberikan tutorial singkat lebih lanjut tentang penggunaan fungsi pencarian di halaman web NCBI yang telah ditulis oleh tim kami untuk informasi tambahan.
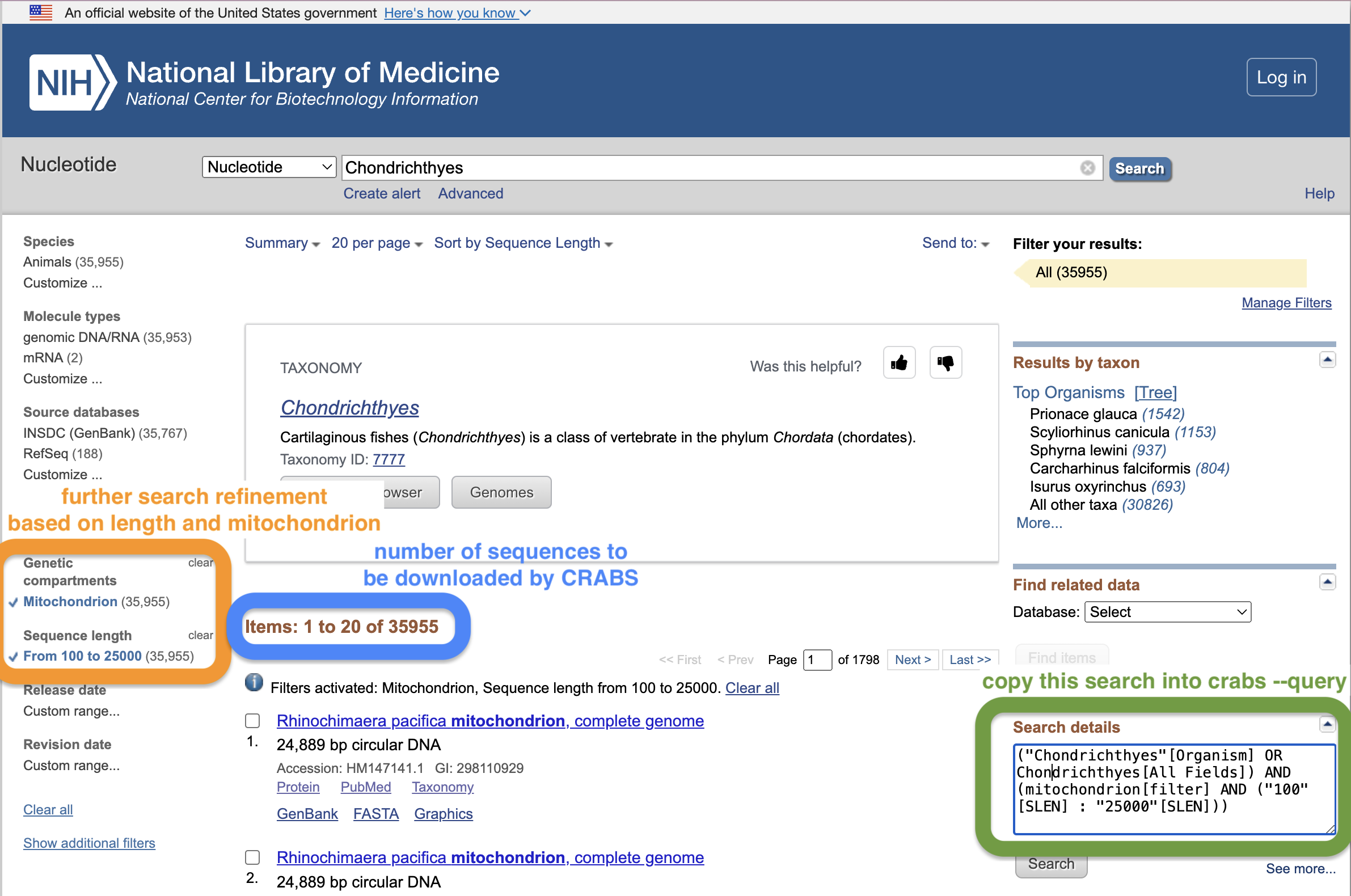
Selain kueri penelusuran ( --query ), pengguna dapat membatasi istilah penelusuran lebih lanjut dengan mengunduh data urutan daftar spesies menggunakan parameter --species . Parameter --species mengambil string masukan nama spesies yang dipisahkan dengan + atau masukan file .txt dengan satu nama spesies per baris dalam dokumen. Parameter --batchsize memberi pengguna opsi untuk mengunduh urutan dalam batch N dari situs web NCBI. Parameter ini defaultnya adalah 5.000. Tidak disarankan untuk meningkatkan nilai ini di atas 5.000, karena server NCBI kemungkinan besar akan memutuskan sambungan unduhan jika terlalu banyak urutan yang diunduh sekaligus. Parameter --email memungkinkan pengguna untuk menentukan alamat email mereka, yang diperlukan untuk mengakses server NCBI. Terakhir, direktori keluaran dan nama file dapat ditentukan menggunakan parameter --output .
crabs --download-ncbi --query '("Chondrichthyes"[Organism] OR Chondrichthyes[All Fields]) AND (mitochondrion[filter] AND ("100"[SLEN] : "25000"[SLEN]))' --output crabs_testing/ncbi_chondrichthyes.fasta --email [email protected] --database nucleotide

--import Setelah data dari repositori online diunduh, file perlu diimpor ke CRABS menggunakan fungsi --import . Format CRABS merupakan satu baris yang dibatasi tab per sekuens yang berisi semua informasi, termasuk (i) ID sekuens, (ii) nama taksonomi yang diurai dari pengunduhan awal, (iii) nomor ID takson NCBI, (iv) garis keturunan taksonomi menurut taksonomi NCBI , dan (v) barisan tersebut. CRABS akan mencoba mendapatkan nomor aksesi NCBI untuk setiap sekuens sebagai ID sekuens. Jika urutan tidak berisi nomor aksesi, yaitu tidak disimpan di NCBI, CRABS akan menghasilkan ID urutan unik menggunakan format berikut: crabs_*[num]*_taxonomic_name . Format dokumen masukan ditentukan menggunakan parameter --import-format dan menentukan nama repositori tempat data diunduh, yaitu BOLD , EMBL , MITOFISH , atau NCBI . Silsilah taksonomi yang dibuat CRABS didasarkan pada taksonomi NCBI dan CRABS memerlukan tiga file yang diunduh menggunakan fungsi --download-taxonomy , yaitu --names , --nodes , dan --acc2tax . Dari versi v 1.0.0 , CRABS mampu menyelesaikan sinonim dan nama yang tidak diterima untuk memasukkan lebih banyak urutan dan keragaman dalam database referensi lokal. Peringkat taksonomi yang akan dimasukkan dalam garis keturunan taksonomi dapat ditentukan menggunakan parameter --ranks . Meskipun peringkat taksonomi apa pun dapat dimasukkan, kami merekomendasikan penggunaan masukan berikut untuk memasukkan semua informasi yang diperlukan untuk sebagian besar pengklasifikasi taksonomi --ranks 'superkingdom;phylum;class;order;family;genus;species' . File keluaran dapat ditentukan menggunakan parameter --output dan merupakan file .txt sederhana. Di Jendela Terminal, CRABS mencetak hasil jumlah sekuens yang diimpor, serta sekuens apa pun yang tidak dapat menghasilkan garis keturunan taksonomi.
crabs --import --import-format bold --input crabs_testing/bold_Elasmobranchii.fasta --names crabs_testing/names.dmp --nodes crabs_testing/nodes.dmp --acc2tax crabs_testing/nucl_gb.accession2taxid --output crabs_testing/crabs_bold.txt --ranks 'superkingdom;phylum;class;order;family;genus;species'

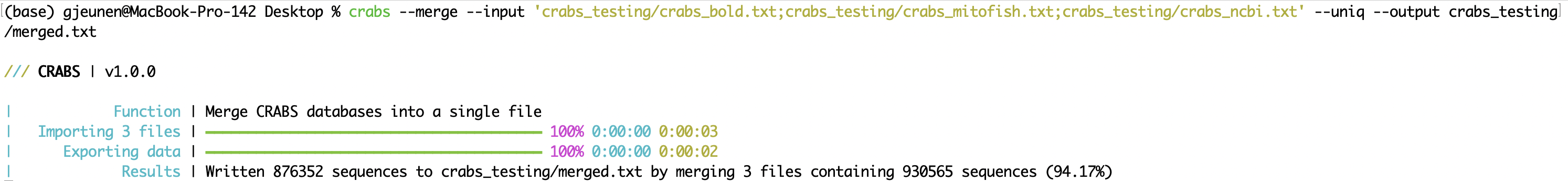
--merge Ketika data urutan dari beberapa repositori online diunduh, file dapat digabungkan menjadi satu file setelah diimpor (lihat 5.2.1 --import ) menggunakan fungsi --merge . File input yang akan digabungkan dapat dimasukkan menggunakan parameter --input , dengan file dipisahkan oleh ; . Ada kemungkinan bahwa suatu urutan diunduh beberapa kali ketika disimpan di berbagai repositori online. Penggunaan parameter --uniq hanya mempertahankan satu versi dari setiap nomor aksesi. File keluaran dapat ditentukan menggunakan parameter --output . Di Jendela Terminal, CRABS mencetak hasil jumlah urutan yang digabungkan, serta jumlah urutan yang dipertahankan saat menggunakan parameter --uniq .
crabs --merge --input 'crabs_testing/crabs_bold.txt;crabs_testing/crabs_mitofish.txt;crabs_testing/crabs_ncbi.txt' --uniq --output crabs_testing/merged.txt
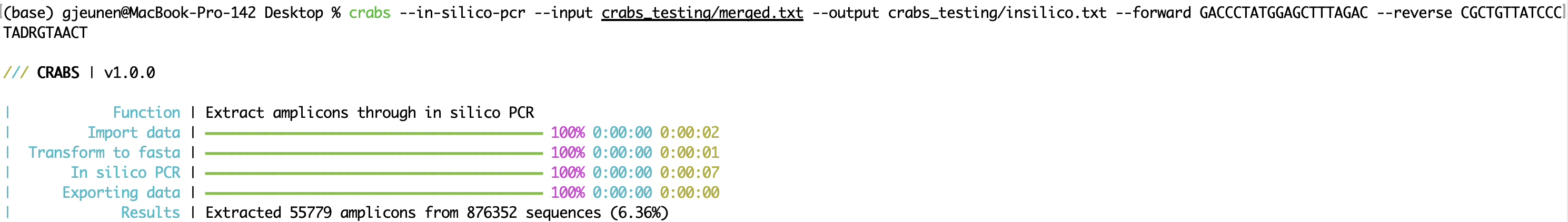
CRABS mengekstrak wilayah amplikon dari set primer dengan melakukan PCR in silico (fungsi: --in-silico-pcr ). CRABS menggunakan cutadapt v 4.4 untuk PCR in silico guna meningkatkan kecepatan eksekusi kode python tradisional. Nama file masukan dan keluaran dapat ditentukan masing-masing menggunakan parameter ' --input ' dan ' --output '. Primer maju dan mundur harus disediakan dalam arah 5'-3' menggunakan parameter ' --forward ' dan ' --reverse '. CRABS akan melengkapi reverse primer. Mulai versi v 1.0.0 , CRABS mampu mempertahankan barcode di kedua arah menggunakan analisis PCR in silico tunggal. Oleh karena itu, tidak ada langkah komplemen balik dan pengujian ulang PCR in silico yang dilakukan, sehingga meningkatkan kecepatan eksekusi secara signifikan. Untuk mempertahankan urutan yang tidak dapat ditemukan daerah pengikatan primer, file keluaran dapat ditentukan untuk parameter --untrimmed . Jumlah ketidakcocokan maksimum yang diperbolehkan yang ditemukan di daerah pengikatan primer dapat ditentukan menggunakan parameter --mismatch , dengan pengaturan default 4. Terakhir, analisis PCR in silico dapat dilakukan secara multithread di CRABS. Secara default, jumlah maksimum thread yang digunakan, namun pengguna dapat menentukan jumlah thread yang akan digunakan dengan parameter --threads .
crabs --in-silico-pcr --input crabs_testing/merged.txt --output crabs_testing/insilico.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT
Merupakan praktik umum untuk menghapus daerah pengikatan primer dari urutan referensi ketika disimpan dalam database online. Oleh karena itu, ketika urutan referensi dihasilkan menggunakan primer maju dan/atau mundur yang sama seperti yang dicari dalam fungsi --in-silico-pcr , fungsi --in-silico-pcr akan gagal memulihkan wilayah amplikon dari urutan referensi. Untuk memperhitungkan kemungkinan ini, CRABS memiliki opsi untuk menjalankan Penyelarasan Global Berpasangan, yang diimplementasikan menggunakan VSEARCH v 2.16.0 , untuk mengekstrak wilayah amplikon yang urutan referensinya tidak berisi wilayah pengikatan primer maju dan mundur secara penuh. Untuk mencapai hal ini, fungsi --pairwise-global-alignment mengambil file database yang diunduh menggunakan parameter --input . Basis data yang akan dicari adalah file keluaran dari --in-silico-pcr dan dapat ditentukan menggunakan parameter --amplicons . File keluaran dapat ditentukan menggunakan parameter --output . Urutan primer, hanya digunakan untuk menghitung panjang pasangan basa, dapat diatur dengan parameter --forward dan --reverse . Karena fungsi --pairwise-global-alignment membutuhkan waktu lama untuk dijalankan pada database besar, panjang urutan dapat dibatasi untuk mempercepat proses menggunakan parameter --size-select . Persentase minimum identitas dan cakupan kueri dapat ditentukan menggunakan parameter --percent-identity dan --coverage . --percent-identity harus diberikan dalam nilai persentase antara 0 dan 1 (misalnya, 95% = 0,95), sedangkan --coverage harus diberikan dalam nilai persentase antara 0 dan 100 (misalnya, 95% = 95). Secara default, fungsi --pairwise-global-alignment dibatasi untuk mempertahankan rangkaian di mana rangkaian primer tidak sepenuhnya ada dalam rangkaian referensi (penyelarasan dimulai atau diakhiri dalam panjang primer maju atau mundur). Ketika parameter --all-start-positions disediakan, hit positif akan disertakan ketika penyelarasan ditemukan di luar rentang daerah pengikatan primer (dilewatkan oleh fungsi --in-silico-pcr karena terlalu banyak ketidakcocokan dalam daerah pengikatan primer). Kami tidak menyarankan penggunaan --all-start-positions , karena sangat kecil kemungkinannya kode batang akan diperkuat menggunakan rangkaian primer yang ditentukan dari fungsi --in-silico-pcr ketika terdapat lebih dari 4 ketidakcocokan pada primer- wilayah yang mengikat.
crabs --pairwise-global-alignment --input crabs_testing/merged.txt --amplicons crabs_testing/insilico.txt --output crabs_testing/aligned.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT --size-select 10000 --percent-identity 0.95 --coverage 0.95
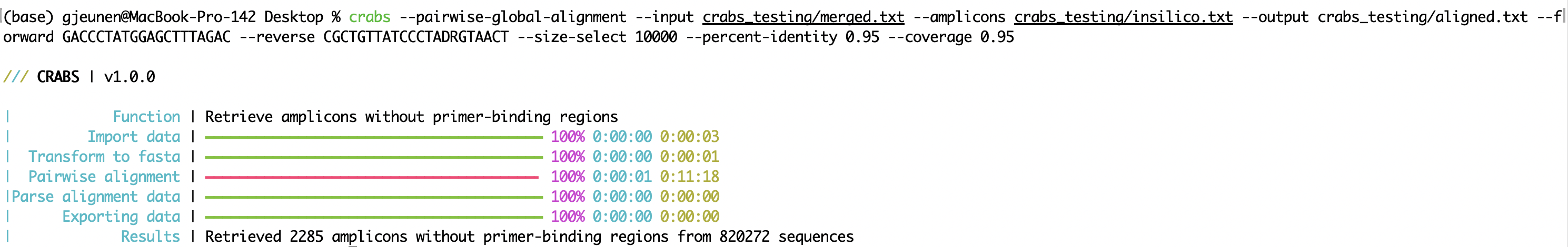
--pairwise-global-alignment Fungsi --pairwise-global-alignment memerlukan banyak waktu untuk dijalankan ketika CRABS memproses file urutan besar, meskipun multithreading didukung. Sejak pembaruan ke CRABS v 1.0.0 , struktur file yang identik telah diterapkan dari --import hingga --export , sehingga memungkinkan fungsi dijalankan dalam urutan apa pun. Meskipun kami tetap menyarankan untuk mengikuti urutan alur kerja CRABS, fungsi --pairwise-global-alignment dapat dipercepat secara signifikan saat menjalankan fungsi --dereplicate dan --filter sebelum fungsi --in-silico-pcr . Dengan menjalankan langkah-langkah kurasi ini sebelum --in-silico-pcr , jumlah urutan yang perlu diproses oleh CRABS untuk fungsi --pairwise-global-alignment akan berkurang secara signifikan.
CATATAN 1 : saat menjalankan fungsi --filter sebelum --in-silico-pcr , pastikan untuk menghilangkan parameter apa pun yang berdampak langsung pada rangkaian, karena --filter akan mendasarkan ini pada keseluruhan rangkaian dan bukan amplikon yang diekstraksi . Oleh karena itu, hilangkan parameter berikut: --minimum-length , --maximum-length , --maximum-n .
CATATAN 2 : ketika menjalankan fungsi --dereplicate dan --filter sebelum --in-silico-pcr , disarankan untuk menjalankan kedua fungsi tersebut lagi setelah --pairwise-global-alignment , karena database sekarang dapat dikurasi lebih lanjut bahwa amplikon diekstraksi.
Setelah semua kode batang potensial untuk set primer telah diekstraksi oleh fungsi --in-silico-pcr dan --pairwise-global-alignment , database referensi lokal dapat menjalani kurasi dan subsetting lebih lanjut dalam CRABS menggunakan berbagai fungsi, termasuk --dereplicate , --filter , dan --subset .
--dereplicate Metode kurasi pertama adalah dengan membatalkan replikasi database referensi lokal menggunakan fungsi --dereplicate . Ada kemungkinan bahwa untuk taksa tertentu, beberapa kode batang identik terdapat dalam database referensi lokal pada saat ini. Hal ini dapat terjadi ketika kelompok penelitian yang berbeda telah menyimpan sekuens yang identik atau jika variasi intra-spesifik antar sekuens untuk suatu takson tidak terkandung dalam kode batang yang diekstraksi. Yang terbaik adalah menghapus kode batang referensi yang identik ini untuk mempercepat penetapan taksonomi, serta meningkatkan hasil penetapan taksonomi (terutama untuk pengklasifikasi taksonomi yang memberikan hasil dalam jumlah terbatas, yaitu BLAST).
File masukan dan keluaran dapat ditentukan masing-masing menggunakan parameter --input dan --output . CRABS menawarkan tiga metode dereplikasi, yang dapat ditentukan menggunakan parameter --dereplication-method , termasuk:
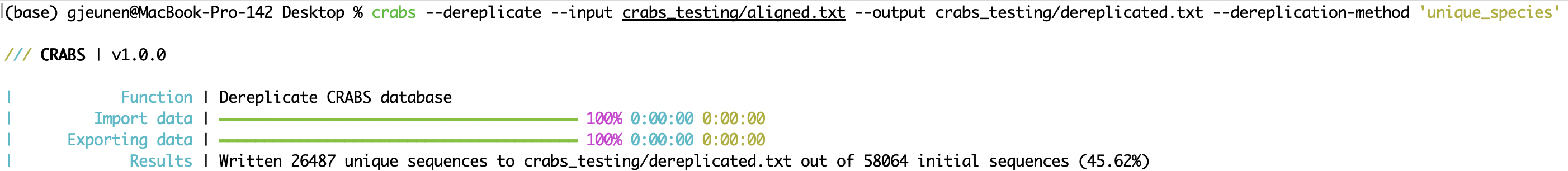
crabs --dereplicate --input crabs_testing/aligned.txt --output crabs_testing/dereplicated.txt --dereplication-method 'unique_species'
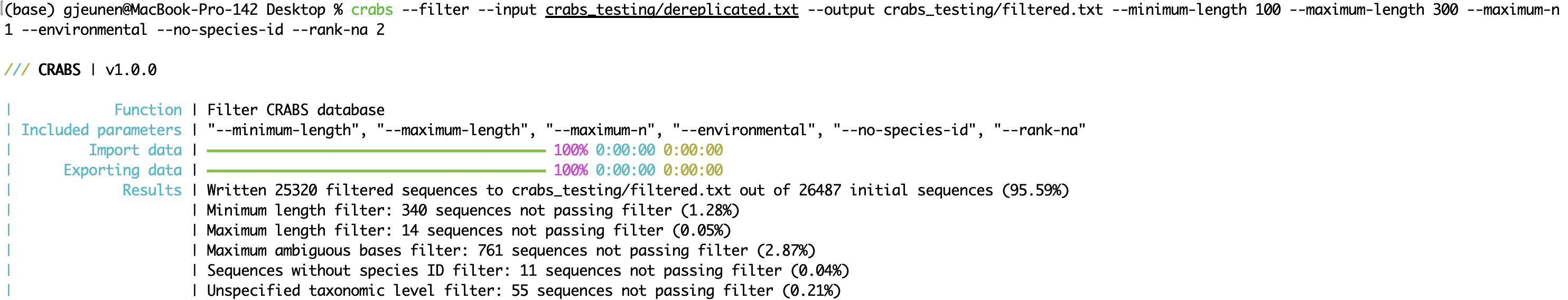
--filter Metode kurasi kedua adalah memfilter database referensi lokal menggunakan berbagai parameter menggunakan fungsi --filter . File masukan dan keluaran dapat ditentukan masing-masing menggunakan parameter --input dan --output . Dari versi v 1.0.0 . CRABS menggabungkan pemfilteran berdasarkan enam parameter, termasuk:
--minimum-length : panjang urutan minimum agar amplikon dapat disimpan dalam database;--maximum-length : panjang urutan maksimum untuk amplikon yang akan disimpan dalam database;--maximum-n : membuang amplikon dengan N atau lebih basis ambigu ( N );--environmental : membuang urutan lingkungan dari database;--no-species-id : membuang urutan yang tidak tersedia nama spesiesnya;--rank-na : membuang urutan dengan N atau lebih tingkat taksonomi yang tidak ditentukan. crabs --filter --input crabs_testing/dereplicated.txt --output crabs_testing/filtered.txt --minimum-length 100 --maximum-length 300 --maximum-n 1 --environmental --no-species-id --rank-na 2
--subset Metode kurasi ketiga dan terakhir yang dimasukkan dalam CRABS adalah dengan membuat subset database referensi lokal untuk menyertakan (parameter: --include ) atau mengecualikan (parameter: --exclude ) taksa tertentu menggunakan fungsi --subset . Fungsi ini memungkinkan penghapusan kode batang referensi dari kelompok taksonomi yang tidak tertarik pada pertanyaan penelitian. Kelompok taksonomi ini dapat dimasukkan ke dalam database referensi lokal karena potensi amplifikasi set primer yang tidak spesifik. Kasus penggunaan lain untuk --subset adalah menghapus urutan kesalahan yang diketahui.
Untuk pengklasifikasi taksonomi berdasarkan pembelajaran mesin (IDTAXA) atau jarak k-mer (SINTAX), akan bermanfaat untuk membuat subset database referensi dengan hanya menyertakan taksa yang diketahui terdapat di wilayah tempat sampel diambil dan mengecualikan spesies berkerabat dekat yang diketahui tidak ada. terjadi di wilayah tersebut untuk meningkatkan resolusi taksonomi yang diperoleh dari pengklasifikasi ini dan memperoleh hasil penugasan taksonomi yang lebih baik.
File masukan dan keluaran dapat ditentukan masing-masing menggunakan parameter --input dan --output . Parameter --include dan --exclude dapat berupa daftar taksa yang dipisahkan oleh ; atau file .txt yang berisi satu nama takson per baris.
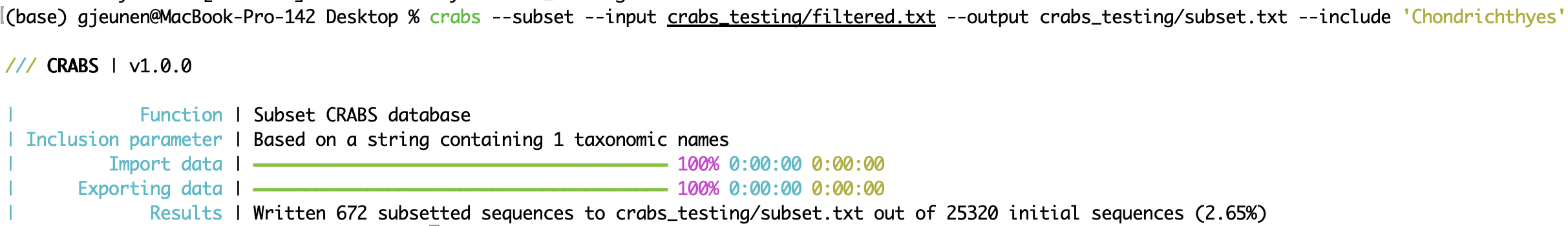
crabs --subset --input crabs_testing/filtered.txt --output crabs_testing/subset.txt --include 'Chondrichthyes'
Setelah database referensi diselesaikan, database tersebut dapat diekspor ke berbagai format untuk mengakomodasi spesifikasi yang dibutuhkan oleh sebagian besar perangkat lunak yang menetapkan taksonomi ke data metagenomik. File masukan dan keluaran dapat ditentukan masing-masing menggunakan parameter --input dan --output . Mulai versi v 1.0.0 , CRABS menggabungkan format database referensi untuk enam pengklasifikasi berbeda (parameter: --export-format ), termasuk:
--export-format 'sintax' : Pengklasifikasi SINTAX digabungkan ke dalam USEARCH dan VSEARCH;--export-format 'rdp' : Pengklasifikasi RDP adalah program mandiri yang banyak digunakan dalam studi mikrobioma;--export-format 'qiime-fasta' dan --export-format 'qiime-text' : Dapat digunakan untuk menetapkan ID taksonomi di QIIME dan QIIME2;--export-format 'dada2-species' dan --export-format 'dada2-taxonomy' : Dapat digunakan untuk menetapkan ID taksonomi di DADA2;--export-format 'idt-fasta' dan --export-format 'idt-text' : Pengklasifikasi IDTAXA adalah algoritme pembelajaran mesin yang tergabung dalam paket DECIPHER R;--export-format 'blast-notax' : Membuat basis data referensi ledakan lokal untuk Blastn dan Megablast di mana output tidak memberikan ID taksonomi, tetapi mencantumkan nomor aksesi;--export-format 'blast-tax' : Membuat basis data referensi ledakan lokal untuk Blastn dan Megablast di mana output menyediakan ID taksonomi dan nomor aksesi. crabs --export --input crabs_testing/subset.txt --output crabs_testing/BLAST_TAX_CHONDRICHTHYES --export-format 'blast-tax'

Saat mengekspor database referensi lokal ke satu format (kecuali untuk pengklasifikasi di mana database referensi dibagi melalui beberapa file, yaitu, qiime, dada2, idtaxa) akan cukup untuk sebagian besar pengguna, loop sederhana dapat ditulis untuk mengekspor lokal Database referensi ke beberapa format jika pengguna ingin membandingkan hasil antara pengklasifikasi taksonomi yang berbeda. Contoh disediakan di bawah ini untuk mengekspor basis data referensi lokal dalam format SINinta, RDP, dan IDTAXA.
for format in sintax.fasta rdp.fasta idt-fasta.fasta idt-text.txt; do crabs --export --input crabs_testing/subset.txt --output crabs_testing/chondrichthyes_${format} --export-format ${format%%.*}; done

Setelah basis data referensi diselesaikan, kepiting dapat menjalankan lima fungsi pasca pemrosesan untuk mengeksplorasi dan memberikan ikhtisar ringkasan dari basis data referensi lokal, termasuk (i) --diversity-figure , (ii) --amplicon-length-figure , ( iii) --phylogenetic-tree , (iv) --amplification-efficiency-figure , dan (v) --completeness-table .
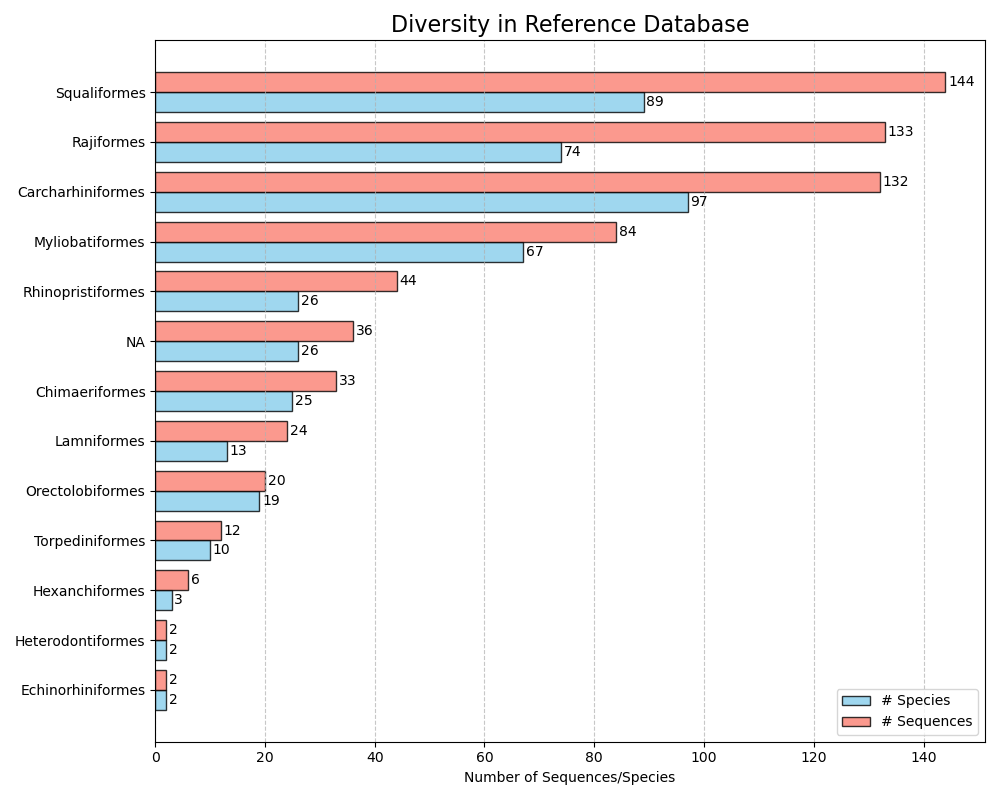
--diversity-figure Fungsi --diversity-figure menghasilkan plot batang horizontal dengan jumlah spesies (berwarna biru) dan jumlah sekuens (dalam oranye) per untuk setiap kelompok taksonomi dalam database referensi. Pengguna dapat menentukan peringkat taksonomi untuk membagi basis data referensi dengan parameter --tax-level . Tingkat pajak adalah jumlah peringkat di mana ia muncul selama fungsi --import . Misalnya, jika --ranks 'superkingdom;phylum;class;order;family;genus;species' digunakan selama-pemisahan --import berdasarkan superkingdom akan membutuhkan --tax-level 1 , filum = --tax-level 2 , class = --tax-level 3 , dll. File input dalam format kepiting dapat ditentukan menggunakan parameter --input . Gambar, dalam format .png, akan ditulis ke file output, yang dapat ditentukan menggunakan parameter --output .
crabs --diversity-figure --input crabs_testing/subset.txt --output crabs_testing/diversity-figure.png --tax-level 4
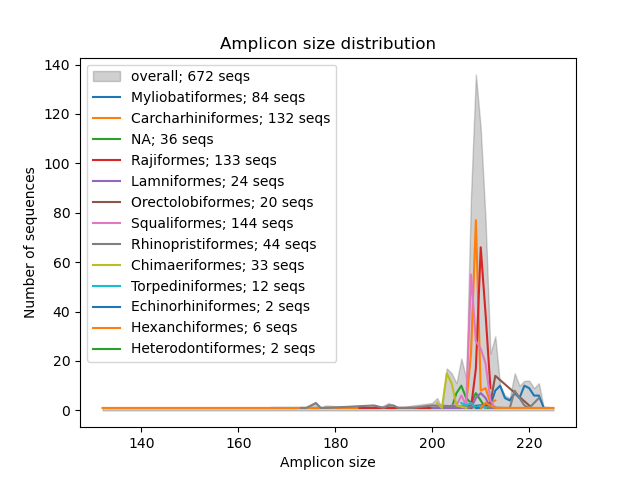
--amplicon-length-figure Fungsi --amplicon-length-figure menghasilkan grafik garis yang menampilkan kisaran panjang amplikon. Kisaran keseluruhan panjang amplikon di semua urutan dalam database referensi ditampilkan dalam warna abu-abu yang diarsir, sedangkan hasilnya terbagi per grup taksonomi (parameter: --tax-level ) ditutup dengan garis berwarna. Selain itu, legenda menampilkan jumlah urutan yang ditetapkan untuk masing -masing kelompok taksonomi dan jumlah total sekuens dalam database referensi. File input dalam format kepiting dapat ditentukan menggunakan parameter --input . Gambar, dalam format .png, akan ditulis ke file output, yang dapat ditentukan menggunakan parameter --output .
crabs --amplicon-length-figure --input crabs_testing/subset.txt --output crabs_testing/amplicon-length-figure.png --tax-level 4
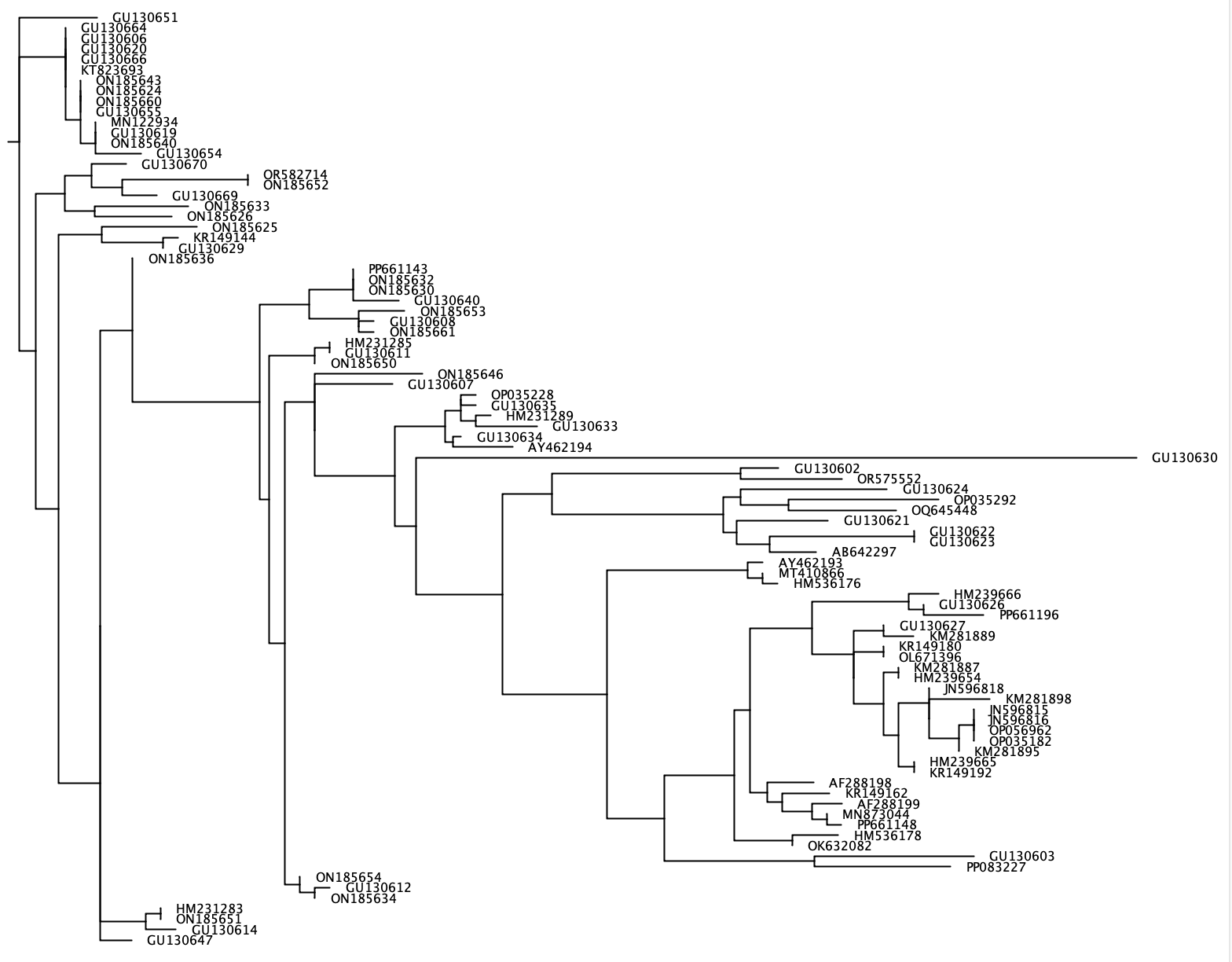
--phylogenetic-tree Fungsi --phylogenetic-tree akan menghasilkan pohon filogenetik untuk daftar spesies yang menarik. Daftar spesies yang menarik ini dapat diimpor menggunakan parameter --species dan terdiri dari salah satu string input yang dipisahkan oleh + atau file .txt dengan satu nama spesies di setiap baris. Untuk setiap spesies yang diminati, urutan akan diekstraksi dari database referensi yang berbagi peringkat taksonomi yang ditentukan pengguna (parameter: --tax-level ) dengan spesies yang diminati. Kepiting akan menghasilkan penyelarasan dari semua urutan yang diekstraksi menggunakan ClustalW2 V 2.1 dan menghasilkan pohon filogenetik yang bergabung dengan tetangga menggunakan FastTree. Pohon filogenetik ini dalam format Newick ini akan ditulis ke file output menggunakan parameter --output dan dapat divisualisasikan dalam program perangkat lunak seperti Figtree atau Geneious. Karena pohon filogenetik terpisah akan dihasilkan untuk setiap spesies yang menarik, --output mengambil nama file generik, sedangkan file output yang tepat akan berisi nama generik ini diikuti oleh '_species_name.tree'.
crabs --phylogenetic-tree --input crabs_testing/subset.txt --output crabs_testing/phylo --tax-level 4 --species 'Carcharodon carcharias+Squalus acanthias'
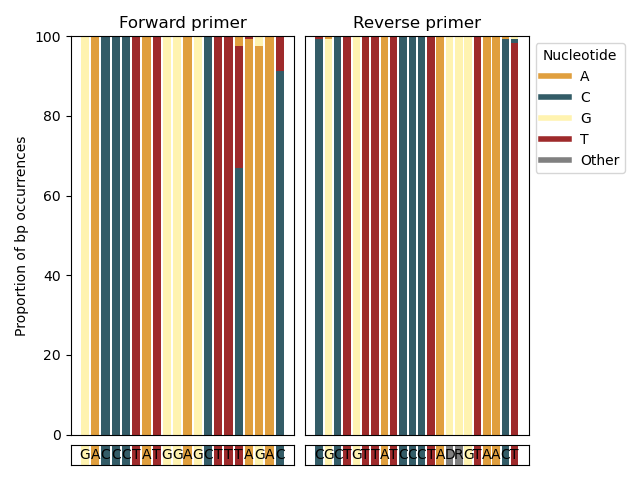
--amplification-efficiency-figure Fungsi --amplification-efficiency-figure akan menghasilkan grafik batang, menampilkan proporsi kejadian pasangan basa di daerah yang mengikat primer untuk kelompok taksonomi yang ditentukan pengguna, sehingga memvisualisasikan tempat-tempat di daerah yang mengikat primer ke depan dan membalikkan ketidakcocokan di mana ketidakcocokan. mungkin terjadi dalam kelompok taksonomi yang diminati, berpotensi mempengaruhi efisiensi amplifikasi. Fungsi --amplification-efficiency-figure mengambil database referensi yang diformat kepiting akhir sebagai input menggunakan parameter --amplicons . Untuk menemukan informasi pada daerah pengikatan primer untuk setiap urutan dalam file input, urutan yang awalnya diunduh setelah impor perlu disediakan menggunakan parameter --input . Urutan primer maju dan terbalik (dalam arah 5 ' -3') disediakan menggunakan parameter --forward dan --reverse . Nama kelompok taksonomi yang diminati dapat disediakan dengan menggunakan parameter --tax-group dan dapat diatur pada tingkat taksonomi yang dimasukkan dalam file input. Akhirnya, gambar dalam format .png akan ditulis ke file output yang ditentukan oleh parameter --output .
crabs --amplification-efficiency-figure --input crabs_testing/merged.txt --amplicons crabs_testing/subset.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT --output crabs_testing/amplification-efficiency.png --tax-group Carcharhiniformes
--completeness-table Fungsi --completeness-table akan menghasilkan tabel yang dibatasi tab (parameter: --output ) dengan informasi tentang daftar spesies yang menarik. Daftar spesies yang menarik ini dapat diimpor menggunakan parameter --species dan terdiri dari salah satu string input yang dipisahkan oleh + atau file .txt dengan nama spesies tunggal pada setiap baris. Silsilah taksonomi akan dihasilkan untuk setiap spesies yang diminati menggunakan file ' name.dmp ' dan ' node.dmp ' yang diunduh menggunakan fungsi --download-taxonomy masing-masing menggunakan parameter --names dan --nodes . Tabel output akan memiliki 10 kolom yang memberikan informasi berikut:
crabs --completeness-table --input crabs_testing/subset.txt --output crabs_testing/completeness.txt --names crabs_testing/names.dmp --nodes crabs_testing/nodes.dmp --species 'Carcharodon carcharias+Squalus acanthias'
crabs --version v 1.0.6 : Perbaikan Bug -> Peningkatan penguraian header tebal selama --import .crabs --version v 1.0.5 : Perbaikan bug -> Menambahkan batasan panjang ke SEQ ID saat membangun basis data ledakan, sesuai kebutuhan untuk perangkat lunak Blast+.crabs --version v 1.0.4 : Info tambahan-> memberikan informasi yang benar tentang input nilai untuk --pairwise-global-alignment --coverage --percent-identity .crabs --version v 1.0.3 : Perbaikan bug -> Memeriksa respons server NCBI 3 kali sebelum membatalkan analisis.crabs --version v 1.0.2 : Bug Fix -> Mampu melaporkan ketika 0 urutan dikembalikan setelah analisis.crabs --version v 1.0.1 : Bug Fix -> Berhasil Membangun Kueri NCBI Menggunakan Parameter --species .


