ClockstaR
1.0.0

セバスチャン・デュシェンヌ、マルティナ・モラック、サイモン・Y・W・ホー。
分子生態・進化・系統学 (MEEP) 研究室
生物科学部
シドニー大学
2015 年 6 月 10 日
partition_data_partitionfinder('drag fasta file with concatenated data here', 'drag partition finder output here')
optim.trees.interactive(folder.parts = 'path to your folder with fasta files and tree topology here')
BSD 距離の導関数を使用してツリー距離の最適化を実装する
トポロジ距離の並列バージョンを実装する
トポロジ距離クラスタリングのチュートリアルを作成する
モデルテスト用にモデルジェネレーターを統合する
RaxML を統合して、分岐の長さとトポロジを最尤最適化する
複数遺伝子データセットを使用して進化のタイムスケールを推定することは、系統発生研究では一般的な作業です。複数遺伝子データセットは、遺伝子、コドン位置、またはその両方によって分割できます。このチュートリアルでは、「データ サブセット」を個々の遺伝子、または複数遺伝子データ セットのサブユニットと呼びます。 「パーティション」という用語は、データ サブセットのグループを指します。
データ サブセットは単一のリラックス クロック モデルで連結して分析できますが、ツリー トポロジが同一であっても、系統間のレート変化のパターンはデータ サブセット間で異なる可能性があります。たとえば、ミトコンドリア遺伝子の系統間の変動率の変動は、核遺伝子の変動率とは異なる場合があります。したがって、進化のタイムスケールと統計的適合性の推定値を改善するために、異なるリラックスクロック モデルを異なるデータ サブセットに割り当てることができます (Duchene and Ho.、2014a を参照)。
複数遺伝子データセットを分割する方法は多数あります。分割スキームを比較する一般的なアプローチは、モデルの適合にベイズ係数または尤度ベースの基準を使用することです。ただし、ほとんどの場合、特にベイズ係数を計算する計算集約的な方法では、考えられるすべての分割スキームをテストすることは不可能です。
ClockstaR は、各データ サブセットの系統分岐の長さを推定します。 sBSDmin として知られる分岐スコア距離は、系統間比率変動パターンの差の尺度として、木のペアごとに計算されます。これらの距離は、パッケージ クラスターに実装されているように、PAM クラスタリング アルゴリズムで GAP 統計を使用して最適なパーティショニング戦略を推論するために使用されます (Maechler et al.、2012) (sBSDmin メトリクスの詳細については、Duchene et al.、2014b を参照) 。
ClockstaR は、複数遺伝子データセットの系統発生的分子時計解析のための R パッケージです。さまざまな遺伝子の系統速度変動のパターンを使用して、クロック分割戦略を選択します。この方法では、系統樹の距離メトリックと教師なし機械学習アルゴリズムを使用して、最適なクロック パートインの数と、各パートティトンでどの遺伝子を分析する必要があるかを特定します。 ClocksktaR で選択された分割ステートジーは、BEAST、MrBayes、PhyloBayes などのプログラムによる後続の分子時計解析に使用できます。
オリジナルの出版物については、このリンクをクリックしてください。
ClockstaR には R のインストールが必要です。また、以下で説明するように、R を通じて取得できるいくつかの R 依存関係も必要です。
ご要望やご質問がございましたら、Sebastian Duchene (sebastian.duchene[at]sydney.edu.au) までお送りください。他のいくつかのソフトウェアとリソースは、シドニー大学の分子生態学、進化、系統発生学研究室で見つけることができます。
このリポジトリを zip ファイルとしてダウンロードし、解凍します。次の手順では、lockstar_example_data フォルダーを使用します。このフォルダーには、いくつかの fasta ファイルと newick 形式の系統樹が含まれています。これらのファイルのいずれかをテキスト ラングラーなどのテキスト エディタで開きます。これらのデータは、進化速度変動の 4 つのパターンに基づいてシミュレートされました。ツリーはすべての遺伝子、またはデータ パーティションのツリー トポロジであることに注意してください。 ClockstaR を実行するには、 Clockstar_example_data のサンプル データと同様にデータをフォーマットしてください。
ClockstaR は GitHub から直接インストールできます。これには devtools パッケージが必要です。 R プロンプトで次のコードを入力して、必要なツールをすべてインストールします (パッケージを直接ダウンロードするにはインターネット接続が必要であることに注意してください)。
install . packages ( " devtools " )
library (devtools)
install_github ( ' ClockstaR ' , ' sebastianduchene ' )ダウンロードしてインストールした後、関数ライブラリを使用して ClockstaR をロードします。
library (ClockstaR2)プログラムの実行方法の例を確認するには、次のように入力します。
example (ClockstaR2)このチュートリアルの残りの部分では、 Clockstar_example_data フォルダーを使用します
最初のステップは、各アライメントの遺伝子ツリーを取得することです。これを行うために、ツリー トポロジーを使用し、個々の遺伝子アライメントのそれぞれ (この場合は A1.fasta から C3.fasta まで) を使用して分岐の長さを最適化します。遺伝子ツリーがある場合は、newick 形式でファイルに保存し、次のステップ (クロックスターを対話的に実行) に進みます。
R プロンプトに次のコードを入力し、Enter キーを押します。
optim . trees . interactive ()パッケージ phangorn のインストールに関するエラー メッセージが表示された場合は、このコードを使用して optim.trees.interactive() を繰り返してください。
install . packcages ( " phangorn " )ClockstaR は次のメッセージを出力します。
Please drag a folder with the data subsets and a tree topology . The files should be in FASTA format, and the trees in NEWICKClockstar_example_data フォルダーを R コンソールにドラッグし、「Enter」と入力します。フォルダーには、FASTA 形式のアリングメントと NEWICK のツリー トポロジのみが含まれている必要があることに注意してください。次のメッセージが表示されます。
What should be the name of the file to save the optimised trees ?最適化されたツリーのファイルの名前を入力します。この場合、「example.trees」を使用します。
example . treesこの時点で、ClockstaR は、遺伝子ごとに個別の置換モデルを使用するか、それともすべての場合に JC を使用するかを尋ねます。これらのデータは JC でシミュレートされたため、「n」を入力して Enter キーを押します。各置換モデルを個別に指定するには、「y」を入力します。
「n」を入力して Enter キーを押すと、ClockstaR が実行を開始します。遺伝子ツリーをグラフィックス デバイスに出力します。指定したツリーがルート化されている場合は、いくつかの警告が出力される場合もありますが、無視しても問題ありません。
Clockstar_example_data フォルダーを開きます。上記のいくつかの手順で指定したように、「example.trees」という名前のファイルが見つかります。テキストエディタで example.trees を開きます。これには、各遺伝子ツリーと、遺伝子アラインメントの名前に応じたツリー名が含まれています。次のようになります。
A1 . fasta (( t1 : 0.01504695462 ,( t2 : 0.00987 ...
A2 . fasta (( t1 : 0.01520523401 ,( t2 : 0.01317 ...
A3 . fasta (( t1 : 0.01519309467 ,( t2 : 0.01092 ...
.
.
.ツリーを含むこのファイルは次のステップで使用されます。
このステップでは、前のステップで取得したような遺伝子ツリーをファイルに保存する必要があります。
上に示すように、R を開いて ClockstaR をロードします。プロンプトで次のコードを入力します。
clockstar . interactive ()ClockstaR は次のメッセージを出力します。
please drag or type in the path to your gene trees file in NEWICK format :遺伝子ツリーを含むファイルを R コンソールにドラッグします。前の手順に従った場合、ファイルの名前は example.trees になります。 Enter を入力します。
インストールしたパッケージによっては、ClockstaR は並行して実行する必要があるかどうかを尋ねる場合があります。これは、大規模なデータ セットの場合に効率的です。ただし、サンプル データの場合は大きな違いはないため、このメッセージが表示された場合は「n」と入力し、次に Enter を入力します。
Packages foreach and doParallel are available for parallel computation
Should we run ClockstaR in parallel (y / n) ? (This is good for large data sets)Clockstar が実行を開始します。画面上の出力は次のようになります。
[ 1 ] " Calculating sBSDmin distances between all pairs of trees "
[ 1 ] " Estimating tree distances "
[ 1 ] " estimating distances 1 of 11 "
[ 1 ] " estimating distances 2 of 11 "
[ 1 ] " estimating distances 3 of 11 "
[ 1 ] " estimating distances 4 of 11 "
[ 1 ] " estimating distances 5 of 11 "
.
.
.木の距離を推定した後 (オリジナルの出版物で説明されています)、ClockstaR は次のメッセージを出力します。
" I finished calculating the sBSDmin distances between trees "
The settings for clustering with ClockstaR are :
PAM clustering algorithm
K from 1 to number of data subsets - 1
SEmax criterion to select the optimal k
500 bootstrap replicates
Are these correct ? (y / n)これらはクラスタリング アルゴリズムの設定です。これらはほとんどのデータ セットに適しているため、この例では「y」を入力して Enter を押すことができます。「n」を入力すると、これらの設定を変更できます。詳細については、Kaufman and Rousseeuw (2009) を参照してください。
ClockstaR はクラスタリング アルゴリズムを実行します。最後に、最適なパーティション数が出力され、結果を PDF ファイルに保存するかどうかを尋ねられます。
[ 1 ] " ClockstaR has finished running "
[ 1 ] " The best number of partitions for your data set is: 3 "
Do you wish to save the results in a pdf file ? (y / n)「y」を入力してEnterします。
次に、ClockstaR は出力ファイルの名前を尋ねます。
What should be the name and path of the output file ?この例では、「example_run」と入力して入力しますが、任意の名前を使用できます。
次に、 Clockstar_example_data フォルダーを開き、2 つの PDF ファイル、example_run_gapstats.pdf と example_run_matrix.pdf を開きます。
example_run_matrix は、FASTA ファイルで名前が付けられているように、行が各遺伝子に対応する行列です。列はパーティションの数を表し、色はクロック パーティションへの各遺伝子の割り当てを表します。たとえば、最適なパーティション数であるk =3 の場合、文字 A、B、および C を持つ遺伝子に個別のクロック パーティションを使用できます。
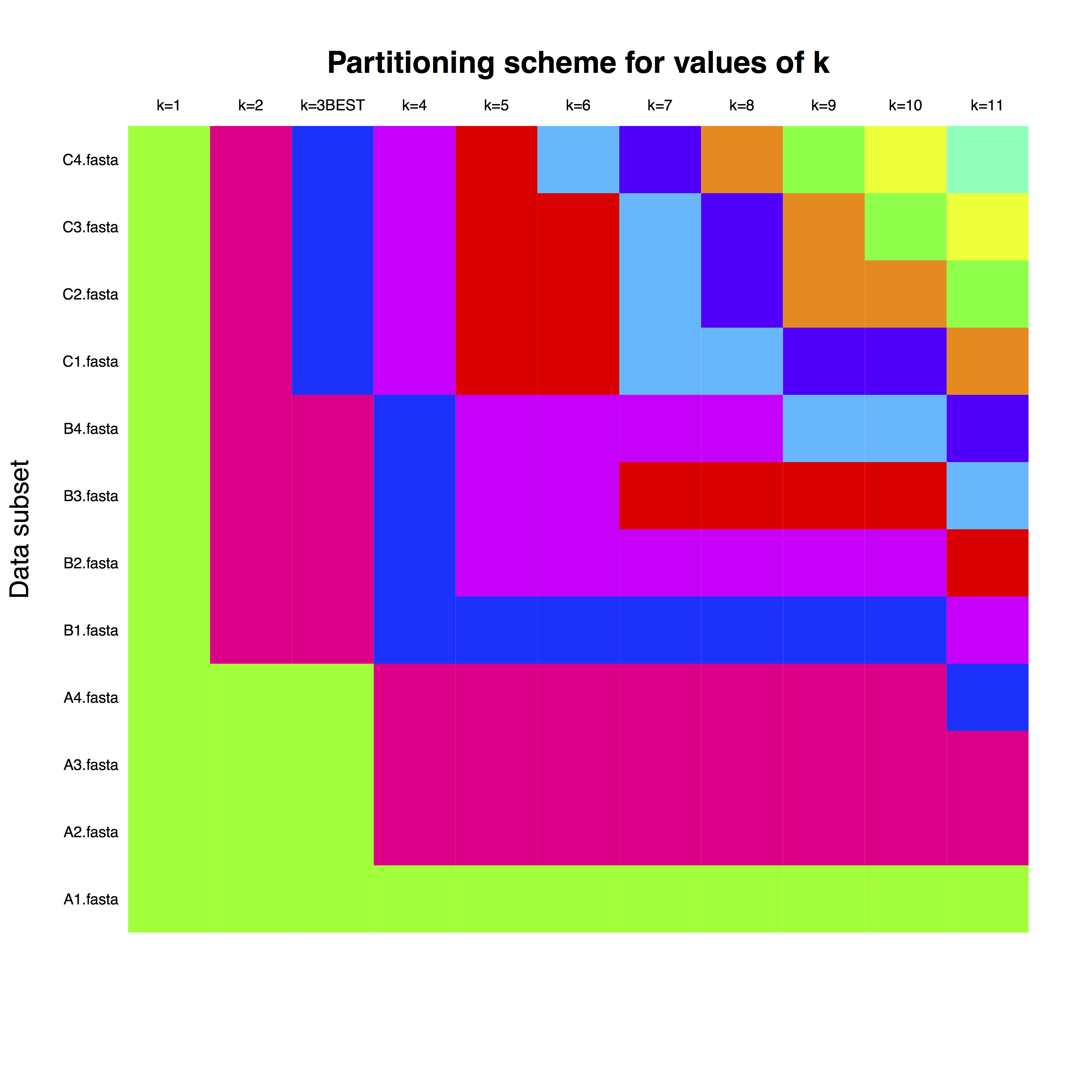
2 番目のプロットは、さまざまな数のパーティションにわたるクラスタリング アルゴリズムの適合です。詳細については、Kaufman and Rousseeuw (2009) およびパッケージ クラスターのドキュメントを参照してください。
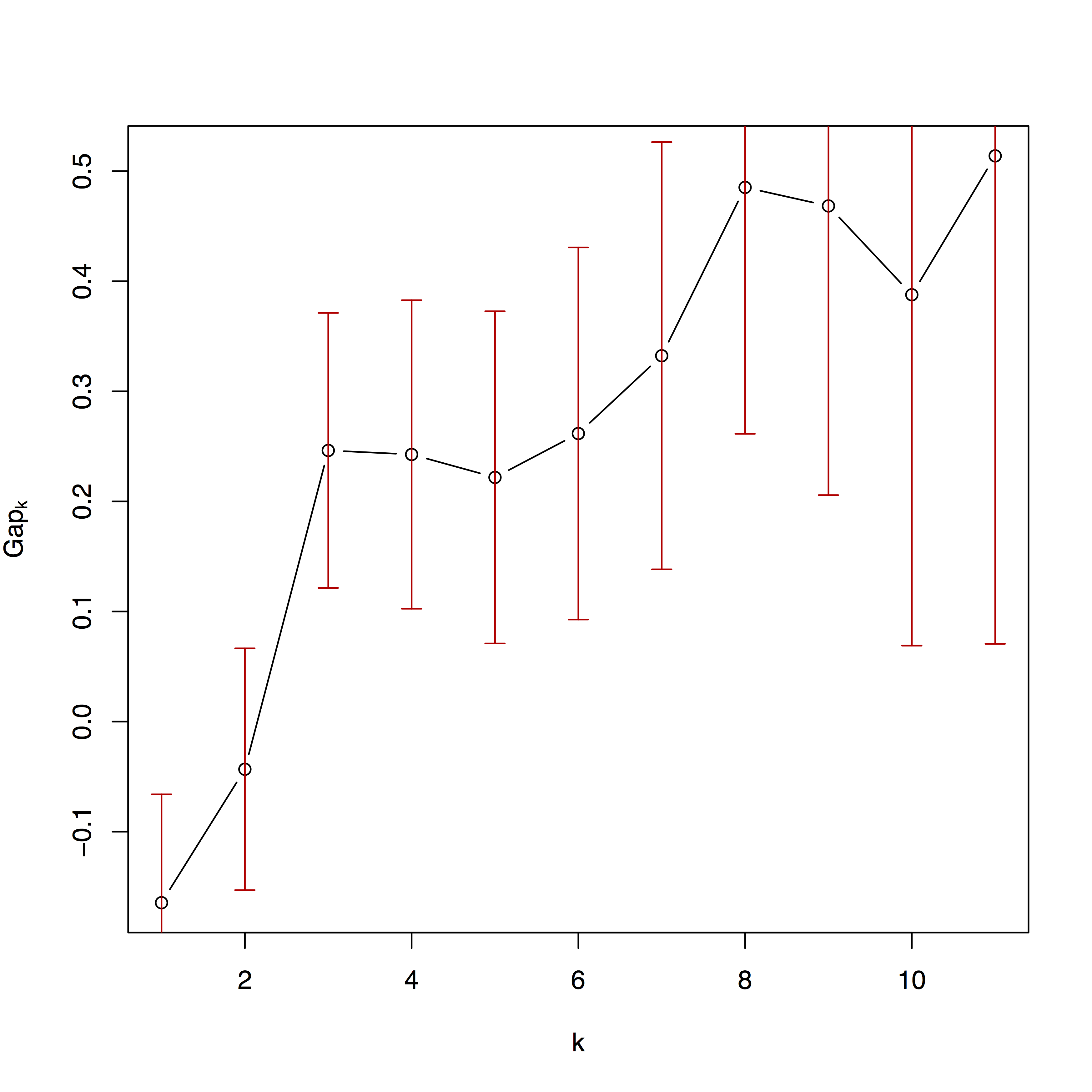
ClockstaR は他のカスタム設定で実行できます。その他の詳細についてはドキュメントを参照するか、ご質問がある場合は sebastian.duchene[at]sydney.edy.au までお問い合わせください。
ロゴはJun Tong氏がデザインしました
デュシェンヌ、S.、ホー、SY (2014a)。複数のリラックスクロックモデルを使用して、DNA 配列データから進化のタイムスケールを推定します。分子系統発生学と進化(77): 65-70。
Duchene、S.、Molak、M.、およびホー、SY (2014b)。 ClockstaR: 分子系統解析におけるリラックスクロック モデルの数の選択。バイオインフォマティクス30 (7): 1017-1019。
カウフマン、L.、ルシーウ、PJ (2009)。データ内のグループを見つける: クラスター分析の概要 (Vol. 344)。ジョン・ワイリー&サンズ。


