このリポジトリには次のものが含まれます。
sepal python3 、できれば 3.5 以降のバージョンが必要です。ダウンロードしてインストールするには、ターミナルを開き、 sepalダウンロードするディレクトリに移動して、次の操作を実行します。
git clone https://github.com/almaan/sepal.git
cd sepal
chmod +x setup.py
./setup.py install
ユーザー権限によっては、 --user引数としてsetup.pyに追加する必要がある場合があります。セットアップを実行すると、拡散時間を計算するために必要な最小限のインストールが行われます。ただし、分析モジュールを使用できるようにするには、推奨パッケージもインストールする必要があります。これを行うには、単に (同じディレクトリ内で) 以下を実行します。
pip install -e " .[full] "繰り返しますが、 --userを含める必要がある場合があります。また、 python-pipインターフェイスをこのように設定している場合は、 pip3使用する必要がある場合があります。 conda環境または仮想環境を使用している場合は、パッケージのインストールに関する推奨事項に従ってください。
これにより、コマンド ライン インターフェイス (CLI) と標準パッケージの両方がインストールされます。インストールが成功したかどうかをテストして確認するには、次のコマンドを実行してみてください。
sepal -h
がく片に関連するヘルプ メッセージを出力する必要があります。ここまでですべてがうまくいった場合は、例のセクションに進み、 sepal動作を確認してください。
sepal の推奨される使用法は、コマンド ライン インターフェイスによるものです。拡散時間を計算するためのシミュレーションとその後の結果の分析または検査は、 sepalと入力してからrunまたはanalyze入力することで簡単に実行できます。 analyzeモジュールには、結果を視覚化する ( inspect )、プロファイルをパターン ファミリに分類する ( family )、または特定されたファミリーを機能強化分析 ( fea ) にかけるなどのさまざまなオプションがあります。使用可能なコマンドの完全なリストを表示するには、 sepal module -hを実行します。ここで、 module はrunおよびanalyzeのいずれかです。以下では、がく片を使用して空間パターンを持つ転写プロファイルを見つける方法を説明します。
結果を保持するフォルダーを作成します。これは作業ディレクトリとしても機能します。リポジトリのメイン ディレクトリから、次の操作を実行します。
cd res
mkdir example
cd exampleMOB サンプルは、分析の例として使用されます。まず、各転写プロファイルの拡散時間を計算します。
sepal run -c ../../data/real/mob.tsv.gz -mo 10 -mc 5 -o . -ar 1以下に、これがどのように表示されるかを示す例を示します (help コマンドの追加表示を含む)。

拡散時間を計算したら、研究と同様に結果を検査したいと思います。上位 20 個のプロファイルを調べます。次のコマンドを実行すると、結果から画像を簡単に生成できます。
sepal analyze -c ../../data/real/mob.tsv.gz
-r 20200409173043610345-top-diffusion-times.tsv
-ar 1k -o . inspect -ng 20 -nc 5これは次の行のようになります。

出力は次の画像になります。
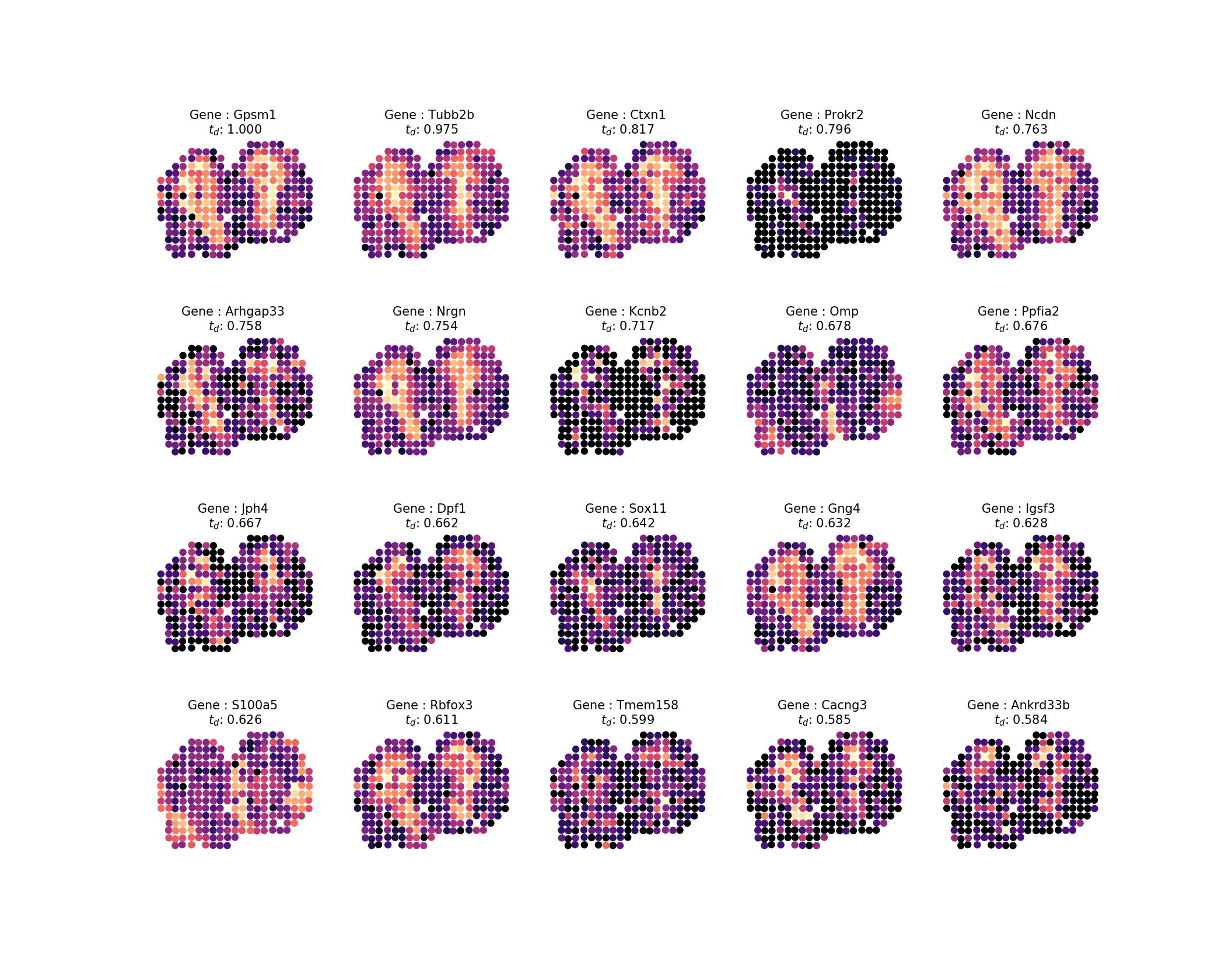
次に、上位 100 個の遺伝子を一連のパターン ファミリーに分類し、パターン内の分散の 85% が固有パターンによって説明されるようにするには、次のようにします。
sepal analyze -c ../../data/real/mob.tsv.gz
-r 20200409173043610345-top-diffusion-times.tsv
-ar 1k -o . family -ng 100 -nbg 100 -eps 0.85 --plot -nc 3これから、各ファミリーの次の 3 つの代表的なモチーフが得られます。
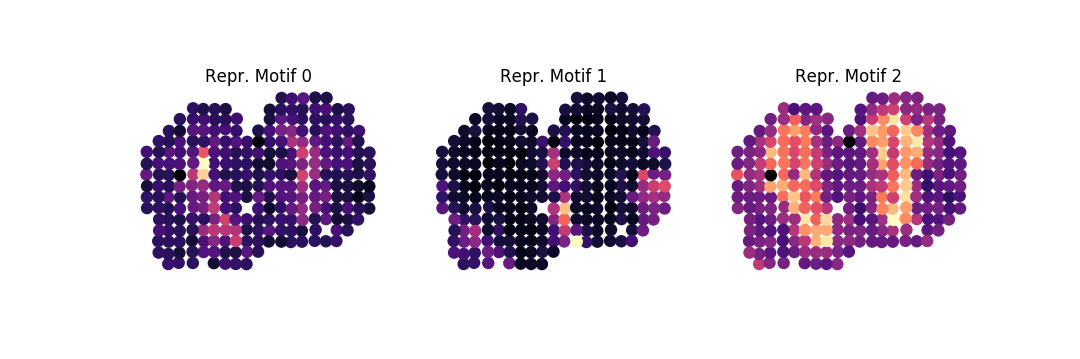
以下を実行することで、家族をエンリッチメント分析の対象にする場合があります。
sepal analyze -c ../../data/real/mob.tsv.gz
-r 20200409173043610345-top-diffusion-times.tsv
-ar 1k -o . fea -fl mob.tsv-family-index.tsv -or " mmusculus "ここで、たとえば、ファミリー 2 では、ニューロンの機能、生成、および制御に関連するいくつかのプロセスが強化されていることがわかります。
| 家族 | ネイティブ | 名前 | p_value | ソース | 交差点のサイズ | |
|---|---|---|---|---|---|---|
| 2 | 2 | GO:0007399 | 神経系の発達 | 0.00035977 | ゴー:BP | 26 |
| 3 | 2 | GO:0050773 | 樹状突起の発達の調節 | 0.000835883 | ゴー:BP | 8 |
| 4 | 2 | GO:0048167 | シナプス可塑性の調節 | 0.00196494 | ゴー:BP | 8 |
| 5 | 2 | GO:0016358 | 樹状突起の発達 | 0.00217167 | ゴー:BP | 9 |
| 6 | 2 | GO:0048813 | 樹状突起の形態形成 | 0.00741589 | ゴー:BP | 7 |
| 7 | 2 | GO:0048814 | 樹状突起の形態形成の制御 | 0.00800399 | ゴー:BP | 6 |
| 8 | 2 | GO:0048666 | ニューロンの発達 | 0.0114088 | ゴー:BP | 16 |
| 9 | 2 | GO:0099004 | カルモジュリン依存性キナーゼシグナル伝達経路 | 0.0159572 | ゴー:BP | 3 |
| 10 | 2 | GO:0050804 | 化学シナプス伝達の調節 | 0.0341913 | ゴー:BP | 10 |
| 11 | 2 | GO:0099177 | トランスシナプスシグナル伝達の調節 | 0.0347783 | ゴー:BP | 10 |
もちろん、この分析は決して網羅的なものではありません。これは、 sepalの CLI を操作する方法を示す簡単な例です。
sepalスタンドアロン ツールとして設計されていますが、機能をインポートして統合ワークフローで使用できる標準の Python パッケージとしても機能するように構築しました。これがどのように行われるかを示すために、黒色腫分析を再現した例を示します。後でさらに例が追加される可能性があります。
sepalへの入力は、 n_locations x n_genes形式である必要がありますが、データが逆の方法で構造化されている場合 ( n_genes x n_locations )、シミュレーションまたは解析の実行時に--transposeフラグを指定するだけで処理されます。の。
現在、 .csv 、 .tsvおよび.h5ad形式をサポートしています。後者の場合、ファイルはこの形式に従って構造化されている必要があります。近い将来、 scanpyチームから空間データの標準化された形式が発表される予定ですが、それまでは前述の標準を使用する予定です。
私たちが使用した実際のデータはすべて公開されており、次のリンクからアクセスできます。
合成データは次のように生成されました。
synthetic/img2cnt.pysynthetic/turing.pysynthetic/ablation.py 研究で示されたすべての結果は、実際のデータと合成データの両方について、 resフォルダーにあります。各サンプルについて、それに応じて結果を構造化しました。
res/sample-name/X-diffusion-times.tsv : ランク付けされたすべての遺伝子の拡散時間analysis/ : 二次分析の出力が含まれます

