reference_database_creator
bug fix --in-silico-pcr --untrimmed
カニ ( C食べるR参照データベースあアンプリコン- BアシードS equencing) は、メタゲノム解析用に厳選された参照データベースを生成する多用途のソフトウェア プログラムです。 CRABS ワークフローは 7 つのモジュールで構成されます。(i) オンライン リポジトリからデータをダウンロードします。 (ii) ダウンロードしたデータを CRABS 形式にインポートします。 (iii)インシリコPCR 解析を通じてアンプリコン領域を抽出する。 (iv)インシリコで抽出されたバーコードとのアラインメントを通じて、プライマー結合領域のないアンプリコンを取得します。 (v) 複数のフィルタリング パラメータを使用してローカル データベースをキュレートし、サブセット化します。 (vi) 分類分類子の要件に従って、ローカル データベースをさまざまな形式でエクスポートします。 (vi) 後処理機能、つまり、ローカル参照データベースの概要を探索して提供するための視覚化。これら 7 つのモジュールは 18 の機能に分割されており、以下で説明します。さらに、18 の関数ごとにコード例が提供されています。最後に、MiFish-E プライマー セット用のローカル サメ参照データベースを構築するチュートリアルが、この README 文書の最後に記載されており、参照用のサンプル スクリプトが提供されています。
CRABS がユーザーのフィードバックに基づいてメジャー アップデートとコードの再設計を行ったことを発表できることを嬉しく思います。これにより、独自のローカル参照データベースを構築する際のユーザー エクスペリエンスが向上することを期待しています。
CRABS v 1.0.0に追加された機能と改善点のリストを以下に示します。
CRABS v 1.0.0 は、この GitHub リポジトリを複製することで手動でダウンロードできるようになりました (詳細については、「4.1 手動インストール」を参照してください)。最新バージョンのインストールを容易にするために、Docker コンテナーと conda パッケージをできるだけ早く更新します。
研究プロジェクトで CRABS を使用する場合は、次の論文を引用してください。
[Jeunen, G.-J., Dowle, E., Edgecombe, J., von Ammon, U., Gemmell, N. J., & Cross, H. (2022). crabs—A software program to generate curated reference databases for metabarcoding sequencing data. Molecular Ecology Resources, 00, 1– 14.](https://doi.org/10.1111/1755-0998.13741)
CRABS は、一般的な Unix/Linux 環境で実行されるコマンドライン専用のツールキットであり、python3 のみで記述されています。ただし、CRABS は Python のsubprocessモジュールを利用して bash 構文でいくつかのコマンドを実行することで、Python 特有の特異性を回避し、実行速度を向上させます。 CRABS をインストールする 3 つの方法を提供します。 CRABS の最新バージョンについては、この GitHub リポジトリを複製し、10 個の依存関係を個別にインストールすることによる手動インストールをお勧めします (すべての依存関係のインストール手順は 4.1 手動インストールで提供されます)。 CRABS は Docker および conda 経由でインストールすることもできます。どちらの方法でも、すべての依存関係が自動的に同時インストールされるため、簡単にインストールできます。 Docker コンテナーと conda パッケージを最新の状態に保つことを目指していますが、特に conda パッケージの場合、最新バージョンへの更新にある程度の遅延が発生する可能性があります。以下に 3 つのアプローチすべての詳細を示します。
手動インストールの場合は、まず CRABS リポジトリのクローンを作成します。この手順では、GitHub がコマンド ラインで利用できる必要があります (GitHub のインストール手順)。
git clone https://github.com/gjeunen/reference_database_creator.git
設定によっては、システム上で CRABS を実行可能にする必要がある場合があります。これは、以下のコードを使用して実現できます。
chmod +x reference_database_creator/crabs
CRABS がインストールされたら、すべての依存関係がインストールされ、グローバルにアクセスできることを確認する必要があります。 CRABS の最新バージョン (バージョンv 1.0.0 ) は Python 3.11.7 (または 3.11.7 と互換性のあるバージョン) で動作し、Python に標準付属していない可能性がある 5 つの Python モジュールと 5 つの外部ソフトウェア プログラムに依存します。すべての依存関係を、インストール手順へのリンクとともに以下にリストします。各モジュールおよびソフトウェア プログラムに提供されているバージョン番号は、CRABS が開発されたものです。ただし、それぞれの互換バージョンを使用することもできます。
Python モジュール:
外部ソフトウェア プログラム:
CRABS とすべての依存関係がインストールされると、以下のコードを使用して OS 全体から CRABS にアクセスできるようになります。
export PATH="/path/to/crabs/folder:$PATH"
/path/to/crabs/folderを、OS 上の GitHub リポジトリ フォルダーへの実際のパス、つまり上記のgit cloneコマンド中に作成されたフォルダーに置き換えます。 exportコードを.bash_profileまたは.bashrc ファイルに追加すると、いつでも CRABS にグローバルにアクセスできるようになります。
Docker は、コンピュータから隔離され、Docker Engine と呼ばれる仮想ホスト オペレーティング システムを通じて実行される「コンテナ」内にソフトウェア アプリケーションを展開できるようにするオープンソース プロジェクトです。仮想マシン上で docker を実行する主な利点は、使用するリソースがはるかに少ないことです。この分離は、Mac、Windows、Linux などのほとんどのオペレーティング システムで Docker コンテナを実行できることを意味します。 Docker Desktop を使用するには、無料アカウントのセットアップが必要な場合があります。このリンクには、 Docker の使用の基本がわかりやすく紹介されています。ここに、 Docker マルチバースを開始して方向付けるためのリンクがあります。
Crabs をコンピュータ上で実行するには 2 つのステップしかありません。まず、Docker Desktop をコンピューターにインストールします。これはほとんどのユーザーにとって無料です。 Mac の手順は次のとおりです。ここでは Windows コンピュータの手順を、ここでは Linux の手順を示します(ほとんどの主要な Linux プラットフォームがサポートされています)。 Docker Desktop をインストールして実行したら (コマンド ラインで docker コマンドを使用するにはデスクトップ アプリケーションが実行されている必要があります)、Crabs イメージを「プル」するだけで準備完了です。
docker pull quay.io/swordfish/crabs:0.1.7
Docker アプリケーションのインストールは簡単ですが、それらのアプリケーションを使用するのは最初は少し難しいかもしれません。開始しやすいように、docker バージョンのカニを使用したコマンド例をいくつか提供しました。これらの例は、このリポジトリの docker_intro フォルダーにあります。これらの例から、参照データベース全体のセットアップを実行して、準備が完了しているはずです。私たちは今後もこれらの例を拡張し、さまざまな状況でこれをテストしていきます。 [問題] タブで質問し、フィードバックを提供してください。
conda パッケージをインストールするには、まず conda をインストールする必要があります。詳細については、このリンクを参照してください。 conda がすでにインストールされている場合は、CRABS をインストールする前にconda update condaを使用して conda ツールを更新することをお勧めします。
conda がインストールされたら、以下の手順に従って CRABS とすべての依存関係をインストールします。コマンドは必ず以下の順序で入力してください。
conda create -n CRABS
conda activate CRABS
conda config --add channels bioconda
conda config --add channels conda-forge
conda install -c bioconda crabs
install コマンドを入力すると、conda はリクエストを処理し (これには 1 分ほどかかる場合があります)、インストールされるすべてのパッケージとプログラムを表示し、確認を求めます。 yを入力してインストールを開始します。これが完了すると、CRABS の準備が整います。
このインストールは Mac および Linux システムでテストされました。 Windows Subsystem for Linux (WSL) ではまだテストしていません。
以下のコードを使用して、CRABS が正常にインストールされているかどうかを確認し、ヘルプ情報を表示します。
crabs -hヘルプ情報では、18 の関数が異なるグループに分割されており、各グループの上部に関数がリストされ、その下に必須およびオプションのパラメーターがリストされています。

CRABS には 7 つのモジュールが含まれており、これらには 18 の機能が組み込まれています。
モジュール 1:オンライン リポジトリからデータをダウンロードする
--download-taxonomy : NCBI 分類情報をダウンロードします。--download-bold : Barcode of Life Database (BOLD) から配列データをダウンロードします。--download-embl : European Nucleotide Archive (ENA; EMBL) から配列データをダウンロードします。--download-mitofish : MitoFish データベースから配列データをダウンロードします。--download-ncbi : 国立バイオテクノロジー情報センター (NCBI) から配列データをダウンロードします。モジュール 2:ダウンロードしたデータを CRABS 形式にインポートする
--import : ダウンロードしたシーケンスまたはカスタム バーコードを CRABS 形式にインポートします。--merge : 異なる CRABS 形式のファイルを 1 つのファイルにマージします。モジュール 3:インシリコPCR 解析によるアンプリコン領域の抽出
--in-silico-pcr : プライマー結合領域を特定して削除することにより、ダウンロードしたデータからアンプリコンを抽出します。モジュール 4:プライマー結合領域を持たないアンプリコンの取得
--pairwise-global-alignment : ダウンロードした配列をインシリコで抽出したバーコードにアラインメントすることにより、プライマー結合領域のないアンプリコンを取得します。モジュール 5:複数のフィルタリング パラメータを使用してローカル データベースをキュレーションおよびサブセット化する
--dereplicate : 重複したシーケンスを破棄します。--filter : 複数のフィルタリング パラメータを介してシーケンスを破棄します。--subset : ローカル データベースをサブセット化して、指定された分類グループを保持または除外します。モジュール 6:ローカル データベースをエクスポートする
--export : 使用する分類分類子の要件に従って、CRABS 形式のデータベースをさまざまな形式にエクスポートします。モジュール 7:ローカル参照データベースを探索し、その概要を提供する後処理機能
--diversity-figure : 参照データベースに含まれる指定されたレベルごとの種および配列グループの数を表示する水平棒グラフを作成します。--amplicon-length-figure : 分類グループごとに分けられたアンプリコンの長さの分布を示す折れ線グラフを作成します。--phylogenetic-tree : ターゲット種リストの参照データベースからバーコードを使用して系統樹を作成します。--amplification-efficiency-figure : プライマー結合領域内のミスマッチを表示する棒グラフを作成します。--completeness-table : 分類グループのバーコードの可用性を含むスプレッドシートを作成します。初期シーケンス データは、(i) BOLD、(ii) EMBL、(iii) MitoFish、および NCBI を含む 4 つのオンライン リポジトリから CRABS によってダウンロードできます。バージョンv 1.0.0以降、各リポジトリからのデータのダウンロードは独自の機能に分割されます。さらに、CRABS は、柔軟性を高め、データのダウンロードが失敗した場合のデバッグを可能にするために、ダウンロード後にデータを自動的にフォーマットしません。
CRABS は、配列データをダウンロードするだけでなく、NCBI 分類情報をダウンロードすることもできます。CRABS は、各配列の分類系統を作成するために CRABS を使用します。
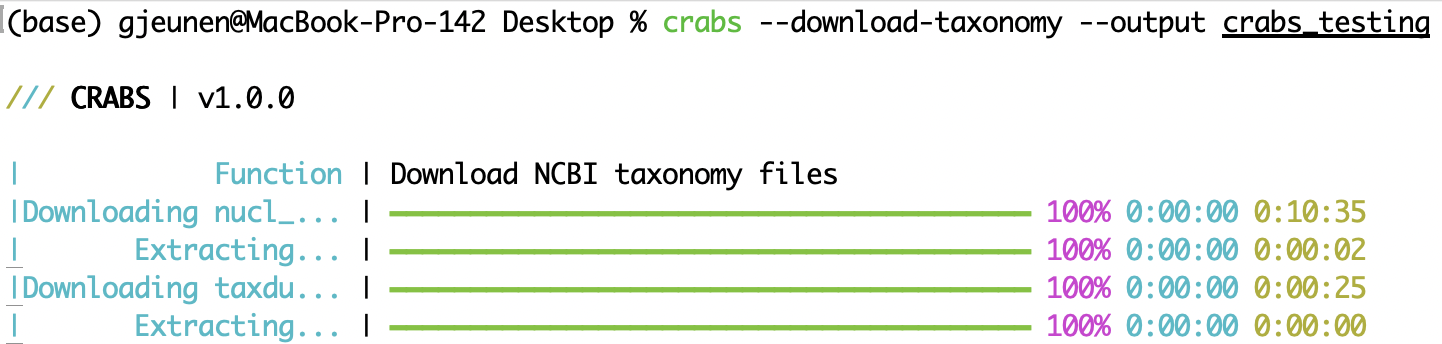
--download-taxonomy参照データベース内のダウンロードされた各配列に分類系統を割り当てるには (5.2 モジュール 2 を参照)、分類情報をダウンロードする必要があります。 CRABS は NCBI の分類を利用し、次の 3 つの特定のファイルをコンピュータにダウンロードします: (i) アクセッション番号を分類 ID にリンクするファイル ( nucl_gb.accession2taxid )、(ii) 各分類 ID に関連付けられた系統名に関する情報を含むファイル ( names.dmp ) )、および (iii) 分類 ID がどのようにリンクされているかに関する情報を含むファイル ( nodes.dmp )。ダウンロードしたファイルの出力ディレクトリは、 --outputパラメータを使用して指定できます。ファイルnucl_gb.accession2taxidまたはファイルnames.dmpおよびnodes.dmpのいずれかを除外するには、 --exclude acc2taxまたは--exclude taxdumpパラメーターをそれぞれ指定できます。 acc2taxとtaxdump両方が--excludeパラメーターに指定されているため、以下の最初のコードはファイルをダウンロードしません。コードの 2 行目は、3 つのファイルすべてをサブディレクトリ--output crabs_testingにダウンロードします。以下のスクリーンショットは、このコード行を実行したときにコンソールに出力される内容を示しています。
crabs --download-taxonomy --exclude 'acc2taxid,taxdump'
crabs --download-taxonomy --output crabs_testing
--download-bold BOLD シーケンスは BOLD Web サイトからダウンロードできます。出力ファイルは 2 行の fasta ドキュメントとして構成されており、 --outputパラメータを使用して指定できます。ユーザーは--taxonパラメーターを使用して、ダウンロードする分類グループを指定できます。ユーザーが複数の分類グループをダウンロードする場合は、単純な for ループ (以下に例を示します) を作成することをお勧めします。これにより、BOLD からインスタンスごとにダウンロードされるデータの量が制限されます。ただし、限られた数の分類グループのみが対象となる場合は、分類グループ名を|で区切ることもできます。 (例を以下に示します)。また、ダウンロードする分類グループ名が太字のアーカイブ内にリストされているかどうか、または別の名前を使用する必要があるかどうかを確認することをお勧めします。たとえば、 --taxon Chondrichthyesを指定しても、このクラス名は太字でリストされていないため、すべての軟骨魚類シーケンスは太字からダウンロードされません。この場合、ユーザーは--taxon Elasmobranchiiを使用する必要があります。ユーザーは、 --markerパラメーターを指定して、ダウンロードを特定の遺伝マーカーに制限するように指定することもできます。複数の遺伝マーカーが対象となる場合は、マーカー名を|で区切る必要があります。 。太字の 4 つの主要な DNA バーコーディング マーカーは、 COI-5P 、 ITS 、 matK 、およびrbcLです。 --markerパラメータの入力では大文字と小文字が区別されます。
推奨されるアプローチ:複数の分類グループの BOLD からデータをダウンロードする単純な for ループ (推奨されるアプローチ)。以下のコードは、最初に Elasmobranchii のデータをダウンロードし、続いて Mammalia に割り当てられたシーケンスをダウンロードします。ダウンロードされたデータは、サブディレクトリ--output crabs_testingに書き込まれ、2 つの別個のファイルに配置され、どのデータがどの分類グループに属するかを示します (つまり、 crabs_testing/bold_Elasmobranchii.fastaとcrabs_testing/bold_Mammalia.fasta )。
for taxon in Elasmobranchii Mammalia; do crabs --download-bold --taxon ${taxon} --output crabs_testing/bold_${taxon}.fasta; done

代替オプション:推奨される for ループのほかに、 |使用して名前を区切ることにより、複数の分類群名を一度に指定できます。 。
crabs --download-bold --taxon 'Elasmobranchii|Mammalia' --output crabs_testing/bold_elasmobranchii_mammalia.fasta
--download-embl EMBL からのシーケンスは、ENA FTP サイトを通じてダウンロードされます。 EMBL ファイルは最初に「.fasta.gz」形式でダウンロードされ、ダウンロードが完了すると自動的に解凍されます。このデータベースは、BOLD や NCBI と比較して、選択的ダウンロードに関してそれほど柔軟性がありません。むしろ、EMBL データは 15 の税区分で構成されており、個別にダウンロードできます。ダウンロードする税区分は、 --taxonパラメーターを使用して指定できます。各税区分は複数のファイルに分割されているため、すべてのファイルをダウンロードするには名前の後に*が付いています。ファイル名を完全に記述することで、特定のファイルをダウンロードすることもできます。 15 の税金分割オプションすべてのリストを以下に示します。出力ディレクトリとファイル名は、 --outputパラメータを使用して指定できます。
税区分のリスト:
crabs --download-embl --taxon 'mam*' --output crabs_testing/embl_mam.fasta
--download-mitofish CRABS では MitoFish データベースをダウンロードすることもできます。このデータベースは 2 行の単一の fasta ファイルです。出力ディレクトリとファイル名は、 --outputパラメータを使用して指定できます。
crabs --download-mitofish --output crabs_testing/mitofish.fasta

--download-ncbi NCBI データベースからのシーケンスは、Entrez プログラミング ユーティリティを通じてダウンロードされます。 NCBI では、ユーザーが--databaseパラメータで指定できるさまざまなデータベースからデータをダウンロードできます。ほとんどのユーザーにとって、ローカル参照データベースを構築するには--database nucleotideデータベースが最も適しています。
NCBI からダウンロードするデータを指定するには、ユーザーは--queryパラメーターを使用して検索を提供します。優れた NCBI 検索を作成するのは難しい場合があります。検索クエリを作成する良い方法は、NCBI Web ページの検索ウィンドウを使用することです。このリンクから、まず最初の検索を実行し、Enter キーを押します。これにより、検索をさらに絞り込むことができる結果ページが表示されます。以下のスクリーンショットでは、配列の長さを 100 ~ 25,000 bp に制限し、ミトコンドリア配列のみを組み込むことで、検索をさらに絞り込みました。ユーザーは、Web サイトの [検索の詳細] ボックスにテキストをコピーして貼り付け、それを引用符で囲んで--queryパラメーターに指定できます。 NCBI Web ページ検索ウィンドウを使用するもう 1 つの利点は、検索クエリに一致する配列の数が Web ページに表示されることです。これは、CRABS によって報告される配列の数と一致する必要があります。この Web ページでは、追加情報のために私たちのチームが作成した NCBI Web ページの検索機能の使用に関するさらに短いチュートリアルを提供します。
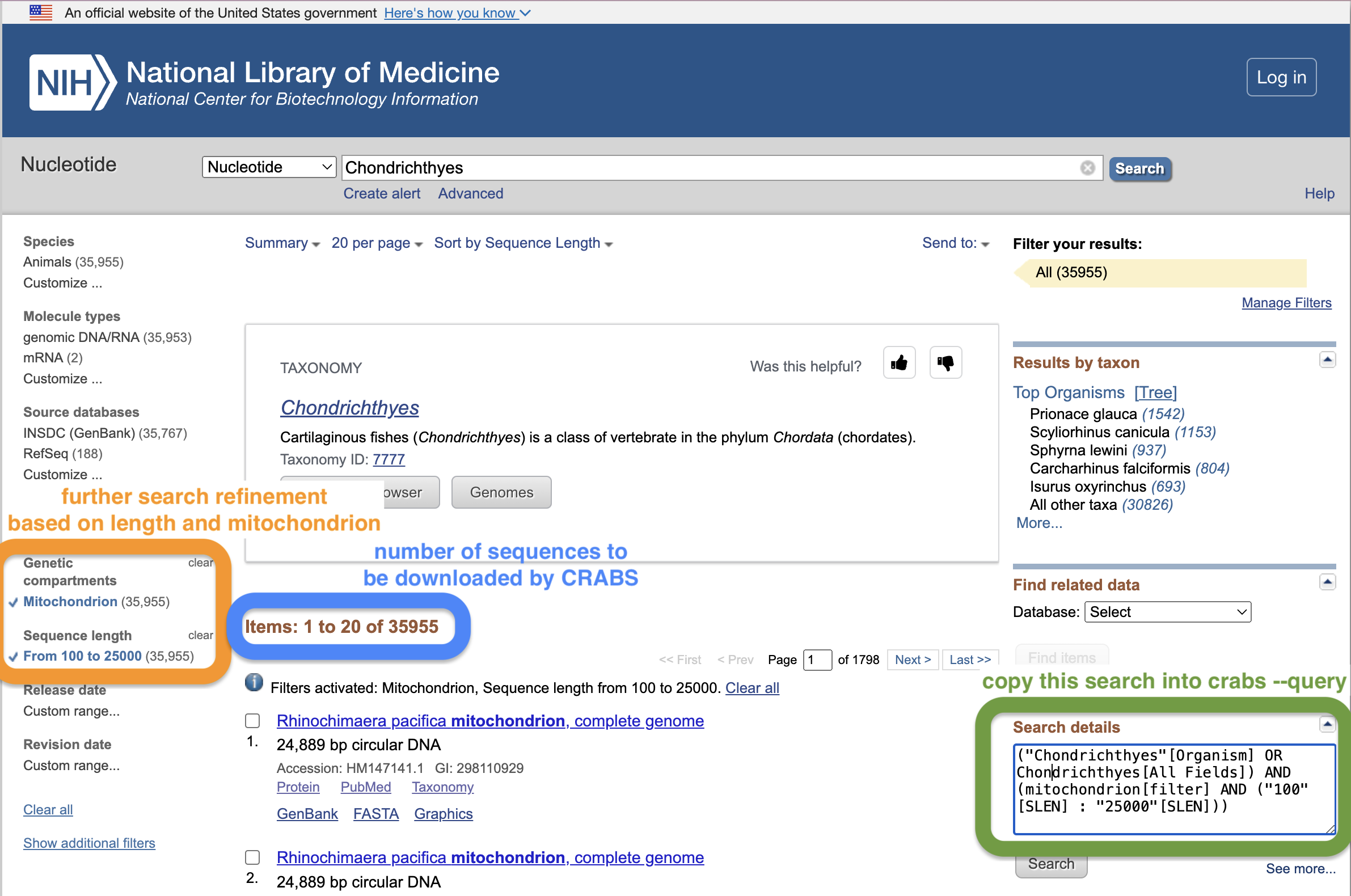
検索クエリ ( --query ) のほかに、ユーザーは--speciesパラメーターを使用して種のリストの配列データをダウンロードすることで、検索用語をさらに制限できます。 --speciesパラメーターは、 +で区切られた種名の入力文字列、またはドキュメント内の 1 行に 1 つの種名が含まれる入力 .txt ファイルを受け取ります。 --batchsizeパラメーターは、NCBI Web サイトから N 個のバッチで配列をダウンロードするオプションをユーザーに提供します。このパラメータのデフォルトは 5,000 です。一度にダウンロードされるシーケンスが多すぎると、NCBI サーバーがダウンロードを切断する可能性が高いため、この値を 5,000 より大きくすることはお勧めできません。 --emailパラメータを使用すると、ユーザーは NCBI サーバーにアクセスするために必要な電子メール アドレスを指定できます。最後に、 --outputパラメータを使用して出力ディレクトリとファイル名を指定できます。
crabs --download-ncbi --query '("Chondrichthyes"[Organism] OR Chondrichthyes[All Fields]) AND (mitochondrion[filter] AND ("100"[SLEN] : "25000"[SLEN]))' --output crabs_testing/ncbi_chondrichthyes.fasta --email [email protected] --database nucleotide

--importオンライン リポジトリからデータをダウンロードしたら、 --import関数を使用してファイルを CRABS にインポートする必要があります。 CRABS 形式は、(i) 配列 ID、(ii) 最初のダウンロードから解析された分類名、(iii) NCBI 分類群 ID 番号、(iv) NCBI 分類法に従った分類系統を含むすべての情報を含む配列ごとにタブ区切りの 1 行で構成されます。 、および (v) シーケンス。 CRABS は、各配列の NCBI アクセッション番号を配列 ID として取得しようとします。配列にアクセッション番号が含まれていない場合、つまり NCBI に寄託されていない場合、CRABS は次の形式を使用して一意の配列 ID を生成します: crabs_*[num]*_taxonomic_name 。入力ドキュメントの形式は、 --import-formatパラメータを使用して指定され、データのダウンロード元のリポジトリの名前 (つまり、 BOLD 、 EMBL 、 MITOFISH 、またはNCBI )を指定します。 CRABS が作成する分類系統は NCBI 分類に基づいており、CRABS では--download-taxonomy関数を使用してダウンロードされた 3 つのファイル (つまり、 --names 、 --nodes 、および--acc2taxが必要です。バージョンv 1.0.0以降、CRABS は同義語と受け入れられない名前を解決して、ローカル参照データベースに多数のシーケンスと多様性を組み込むことができるようになりました。分類系統に含める分類ランクは、 --ranksパラメーターを使用して指定できます。任意の分類学的ランクを含めることができますが、次の入力を使用して、ほとんどの分類学的分類子に必要な情報をすべて含めることをお勧めします--ranks 'superkingdom;phylum;class;order;family;genus;species' 。出力ファイルは--outputパラメーターを使用して指定でき、単純な .txt ファイルです。ターミナル ウィンドウで、CRABS はインポートされた配列の数の結果と、分類学的系統を生成できなかった配列を出力します。
crabs --import --import-format bold --input crabs_testing/bold_Elasmobranchii.fasta --names crabs_testing/names.dmp --nodes crabs_testing/nodes.dmp --acc2tax crabs_testing/nucl_gb.accession2taxid --output crabs_testing/crabs_bold.txt --ranks 'superkingdom;phylum;class;order;family;genus;species'

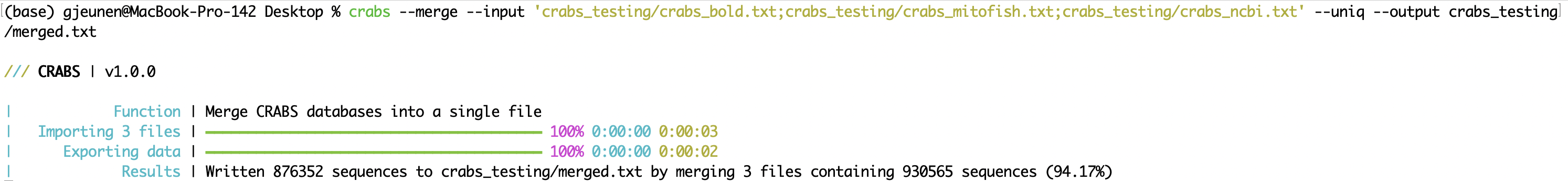
--merge複数のオンライン リポジトリから配列データをダウンロードする場合、 --merge関数を使用してインポート (5.2.1 --import参照) 後にファイルを 1 つのファイルにマージできます。マージする入力ファイルは、 --inputパラメータを使用して入力できます。ファイルは ; で区切られます; 。シーケンスがさまざまなオンライン リポジトリに保存されるときに複数回ダウンロードされた可能性があります。 --uniqパラメータを使用すると、各アクセッション番号の単一バージョンのみが保持されます。出力ファイルは--outputパラメータを使用して指定できます。ターミナル ウィンドウでは、CRABS はマージされたシーケンスの数の結果と、 --uniqパラメーターを使用したときに保持されるシーケンスの数を出力します。
crabs --merge --input 'crabs_testing/crabs_bold.txt;crabs_testing/crabs_mitofish.txt;crabs_testing/crabs_ncbi.txt' --uniq --output crabs_testing/merged.txt
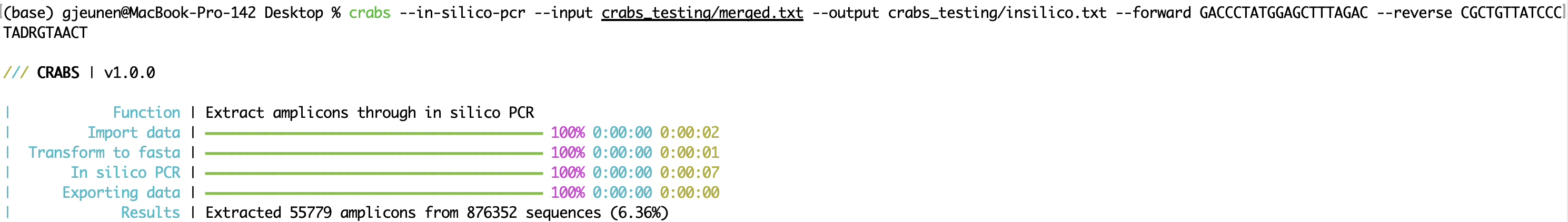
CRABS は、 in silico PCR (関数: --in-silico-pcr ) を実行することにより、プライマーセットのアンプリコン領域を抽出します。 CRABS は、インシリコPCR に Cutadapt v 4.4 を使用して、従来の Python コードの実行速度を向上させます。入力ファイル名と出力ファイル名は、それぞれ「 --input 」パラメータと「 --output 」パラメータを使用して指定できます。フォワードプライマーとリバースプライマーは両方とも、それぞれ「 --forward 」パラメータと「 --reverse 」パラメータを使用して 5'-3' 方向に提供する必要があります。 CRABS はリバースプライマーを逆相補します。バージョンv 1.0.0以降、CRABS は 1 回のインシリコPCR 分析を使用して両方向でバーコードを保持できるようになりました。したがって、逆補完ステップやインシリコPCR の再実行は実行されず、実行速度が大幅に向上します。プライマー結合領域が見つからなかった配列を保持するには、 --untrimmedパラメーターに出力ファイルを指定できます。プライマー結合領域で見つかったミスマッチの最大許容数は、 --mismatchパラメーターを使用して指定でき、デフォルト設定は 4 です。最後に、インシリコPCR 解析を CRABS でマルチスレッド化できます。デフォルトでは、最大数のスレッドが使用されますが、ユーザーは--threadsパラメータで使用するスレッドの数を指定できます。
crabs --in-silico-pcr --input crabs_testing/merged.txt --output crabs_testing/insilico.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT
オンラインデータベースに登録される際、参照配列からプライマー結合領域を除去するのが一般的です。したがって、 --in-silico-pcr関数で検索したのと同じフォワードプライマーおよび/またはリバースプライマーを使用して参照配列が生成された場合、 --in-silico-pcr関数はアンプリコン領域の回復に失敗します。参照配列。この可能性を考慮して、CRABS には、VSEARCH v 2.16.0 を使用して実装されたペアワイズ グローバル アライメントを実行して、参照配列に完全なフォワードおよびリバース プライマー結合領域が含まれていないアンプリコン領域を抽出するオプションがあります。これを実現するために、 --pairwise-global-alignment関数は、 --inputパラメーターを使用して、最初にダウンロードされたデータベース ファイルを取り込みます。検索対象のデータベースは--in-silico-pcrからの出力ファイルであり、 --ampliconsパラメーターを使用して指定できます。出力ファイルは--outputパラメータを使用して指定できます。プライマー シーケンスは、塩基対の長さを計算するためにのみ使用され、 --forwardおよび--reverseパラメーターを使用して設定できます。 --pairwise-global-alignment関数は大規模なデータベースでは実行に時間がかかる場合があるため、 --size-selectパラメーターを使用してシーケンスの長さを制限してプロセスを高速化できます。最小パーセント ID とクエリ カバレッジは、それぞれ--percent-identityと--coverageパラメータを使用して指定できます。 --percent-identity 0 ~ 1 (例: 95% = 0.95) のパーセンテージ値として指定する必要があり、 --coverage 0 ~ 100 (例: 95% = 95) のパーセンテージ値として指定する必要があります。デフォルトでは、 --pairwise-global-alignment関数は、プライマー配列が参照配列に完全には存在しない配列 (フォワードまたはリバース プライマーの長さ内で開始または終了するアラインメント) を保持するように制限されています。 --all-start-positionsパラメーターが指定されている場合、アライメントがプライマー結合領域の範囲外で見つかった場合に、ポジティブ ヒットが含まれます (不一致が多すぎるため、 --in-silico-pcr関数で見逃されます)。プライマー結合領域)。プライマーに 4 つを超えるミスマッチが存在する場合、 --in-silico-pcr関数の指定されたプライマー セットを使用してバーコードが増幅される可能性は非常に低いため、 --all-start-positions使用はお勧めしません。結合領域。
crabs --pairwise-global-alignment --input crabs_testing/merged.txt --amplicons crabs_testing/insilico.txt --output crabs_testing/aligned.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT --size-select 10000 --percent-identity 0.95 --coverage 0.95
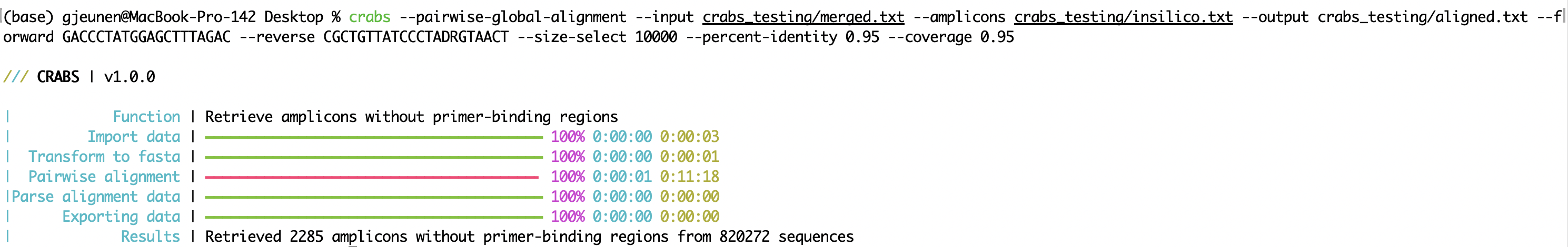
--pairwise-global-alignmentのコード実行を高速化します。 CRABS が大きなシーケンス ファイルを処理している場合、マルチスレッドがサポートされている場合でも、 --pairwise-global-alignment関数の実行にはかなりの時間がかかる可能性があります。 CRABS v 1.0.0への更新以降、 --importから--exportまで同一のファイル構造が導入され、これにより関数を任意の順序で実行できるようになりました。 CRABS ワークフローの順序に従うことをお勧めしますが、 --in-silico-pcr関数の前に--dereplicateと--filter関数を実行すると、 --pairwise-global-alignment関数の速度が大幅に向上する可能性があります。 --in-silico-pcrの前にこれらのキュレーション手順を実行すると、 --pairwise-global-alignment関数のために CRABS によって処理する必要がある配列の数が大幅に減少します。
注 1 : --in-silico-pcrの前に--filter関数を実行する場合、 --filter抽出されたアンプリコンではなく配列全体に基づいているため、配列に直接影響を与えるパラメーターを必ず省略してください。 。したがって、パラメータ--minimum-length 、 --maximum-length 、 --maximum-nを省略します。
注 2 : --in-silico-pcrの前に--dereplicateおよび--filter関数を実行する場合は、データベースをさらに厳選できるため、 --pairwise-global-alignment後に両方の関数を再度実行することをお勧めします。アンプリコンが抽出されること。
--in-silico-pcrおよび--pairwise-global-alignment関数によってプライマー セットの潜在的なバーコードがすべて抽出されると、ローカル参照データベースは、 --dereplicateなどのさまざまな関数を使用して CRABS 内でさらにキュレーションおよびサブセット化できます。 、 --filter 、および--subset 。
--dereplicate最初のキュレーション方法は、 --dereplicate関数を使用してローカル参照データベースを非レプリケートすることです。特定の分類群では、この時点でローカル参照データベース内に複数の同一のバーコードが含まれている可能性があります。これは、異なる研究グループが同一の配列を寄託した場合、または分類群の配列間の種内変異が抽出されたバーコード内に含まれていない場合に発生する可能性があります。分類の割り当てを高速化し、分類の割り当て結果を改善するには、これらの同一の参照バーコードを削除するのが最善です (特に、限られた数の結果を提供する分類分類子、つまり BLAST)。
入力ファイルと出力ファイルは、それぞれ--inputパラメータと--outputパラメータを使用して指定できます。 CRABS は、 --dereplication-methodパラメーターを使用して指定できる次の 3 つのレプリケーション解除方法を提供します。
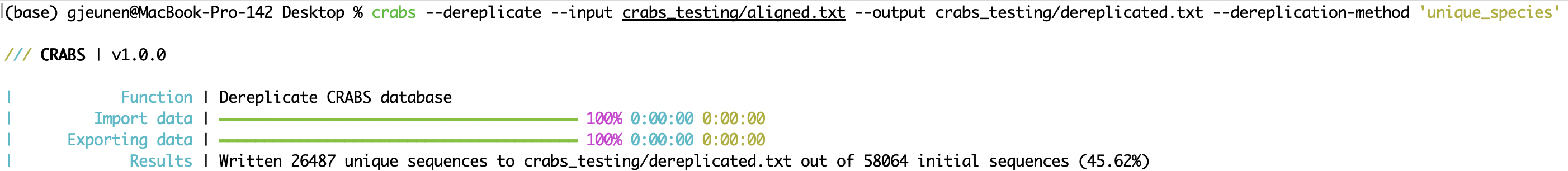
crabs --dereplicate --input crabs_testing/aligned.txt --output crabs_testing/dereplicated.txt --dereplication-method 'unique_species'
--filter 2 番目のキュレーション方法は、 --filter関数を使用してさまざまなパラメーターを使用してローカル参照データベースをフィルター処理することです。入力ファイルと出力ファイルは、それぞれ--inputパラメータと--outputパラメータを使用して指定できます。バージョンv 1.0.0以降。 CRABS には、次の 6 つのパラメータに基づくフィルタリングが組み込まれています。
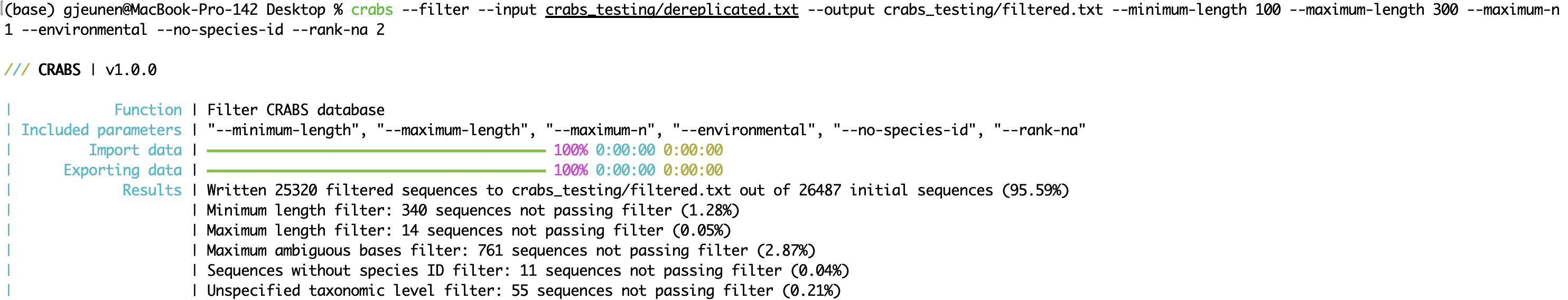
--minimum-length : データベースに保持されるアンプリコンの最小配列長。--maximum-length : データベースに保持されるアンプリコンの最大配列長。--maximum-n : N 個以上のあいまいな塩基 ( N ) を持つアンプリコンを破棄します。--environmental : データベースから環境シーケンスを破棄します。--no-species-id : 種名が利用できないシーケンスを破棄します。--rank-na : N 以上の未指定の分類レベルを持つシーケンスを破棄します。 crabs --filter --input crabs_testing/dereplicated.txt --output crabs_testing/filtered.txt --minimum-length 100 --maximum-length 300 --maximum-n 1 --environmental --no-species-id --rank-na 2
--subset CRABS に組み込まれている 3 番目で最後のキュレーション方法は、 --subset関数を使用して特定の分類群を含めるか (パラメーター: --include )、または除外する (パラメーター: --exclude ) ようにローカル参照データベースをサブセット化することです。この機能により、研究課題に関係のない分類グループから参照バーコードを削除できます。これらの分類群は、プライマー セットの非特異的増幅の可能性があるため、ローカル参照データベースに組み込まれている可能性があります。 --subsetの別の使用例は、既知の誤ったシーケンスを削除することです。
機械学習 (IDTAXA) または k-mer 距離 (SINTAX) に基づく分類分類器の場合、サンプルが採取された地域で発生することがわかっている分類群のみを含め、未確認の近縁種を除外することで参照データベースをサブセット化すると有益な場合があります。これらの分類子の分類学的解像度が向上し、分類法の割り当て結果が向上します。
入力ファイルと出力ファイルは、それぞれ--inputパラメータと--outputパラメータを使用して指定できます。 --includeおよび--excludeパラメーターは、; で区切られた分類群のリストを受け取ることができます;または、1 行に 1 つの分類群名を含む .txt ファイル。
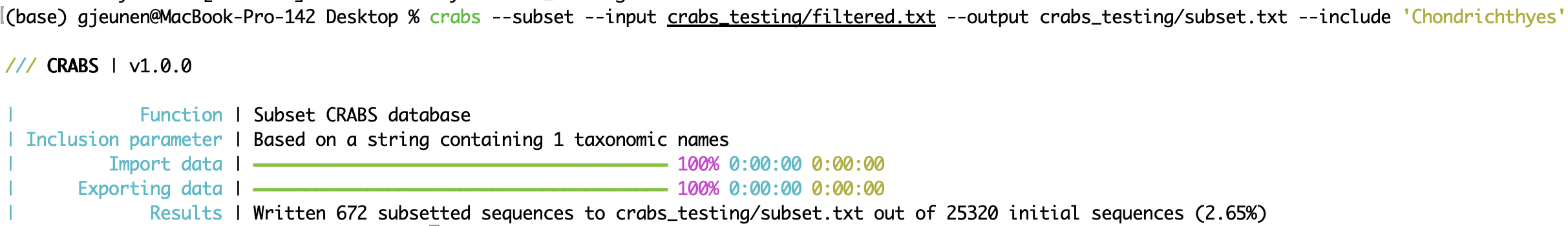
crabs --subset --input crabs_testing/filtered.txt --output crabs_testing/subset.txt --include 'Chondrichthyes'
参照データベースが完成したら、分類法をメタゲノム データに割り当てるほとんどのソフトウェア ツールで必要な仕様に対応するために、さまざまな形式にエクスポートできます。入力ファイルと出力ファイルは、それぞれ--inputパラメータと--outputパラメータを使用して指定できます。バージョンv 1.0.0以降、CRABS には、以下を含む 6 つの異なる分類子 (パラメーター: --export-format ) の参照データベースのフォーマットが組み込まれています。
--export-format 'sintax' : SINTAX 分類子は USEARCH および VSEARCH に組み込まれます。--export-format 'rdp' : RDP 分類子は、マイクロバイオーム研究で広く使用されているスタンドアロン プログラムです。--export-format 'qiime-fasta'および--export-format 'qiime-text' : QIIME および QIIME2 で分類 ID を割り当てるために使用できます。--export-format 'dada2-species'および--export-format 'dada2-taxonomy' : DADA2 で分類 ID を割り当てるために使用できます。--export-format 'idt-fasta'および--export-format 'idt-text' : IDTAXA 分類子は、DECIPHER R パッケージに組み込まれている機械学習アルゴリズムです。--export-format 'blast-notax' :出力が分類IDを提供しないBlastnおよびMegablastのローカルブラストリファレンスデータベースを作成しますが、アクセッション番号をリストします。--export-format 'blast-tax' :BlastnとMegablastのローカルブラストリファレンスデータベースを作成し、出力が分類IDとアクセッション番号の両方を提供します。 crabs --export --input crabs_testing/subset.txt --output crabs_testing/BLAST_TAX_CHONDRICHTHYES --export-format 'blast-tax'

ローカルリファレンスデータベースを単一の形式にエクスポートする(参照データベースが複数のファイル、すなわちQIIME、DADA2、IDTAXAに分割される分類器を除く)、ほとんどのユーザーには十分ですが、LOOPを簡単に記述してローカルをエクスポートできます。ユーザーが異なる分類学者間で結果を比較したい場合、データベースを複数の形式に参照してください。 Sintax、RDP、およびIDTAXA形式のローカルリファレンスデータベースをエクスポートするために、以下に例を示します。
for format in sintax.fasta rdp.fasta idt-fasta.fasta idt-text.txt; do crabs --export --input crabs_testing/subset.txt --output crabs_testing/chondrichthyes_${format} --export-format ${format%%.*}; done

参照データベースが完成すると、CRABSは5つのポストプロセッシング関数を実行して、(i) --diversity-figure 、(ii) --amplicon-length-figure 、( iii) --phylogenetic-tree 、(iv) --amplification-efficiency-figure 、および(v) --completeness-table 。
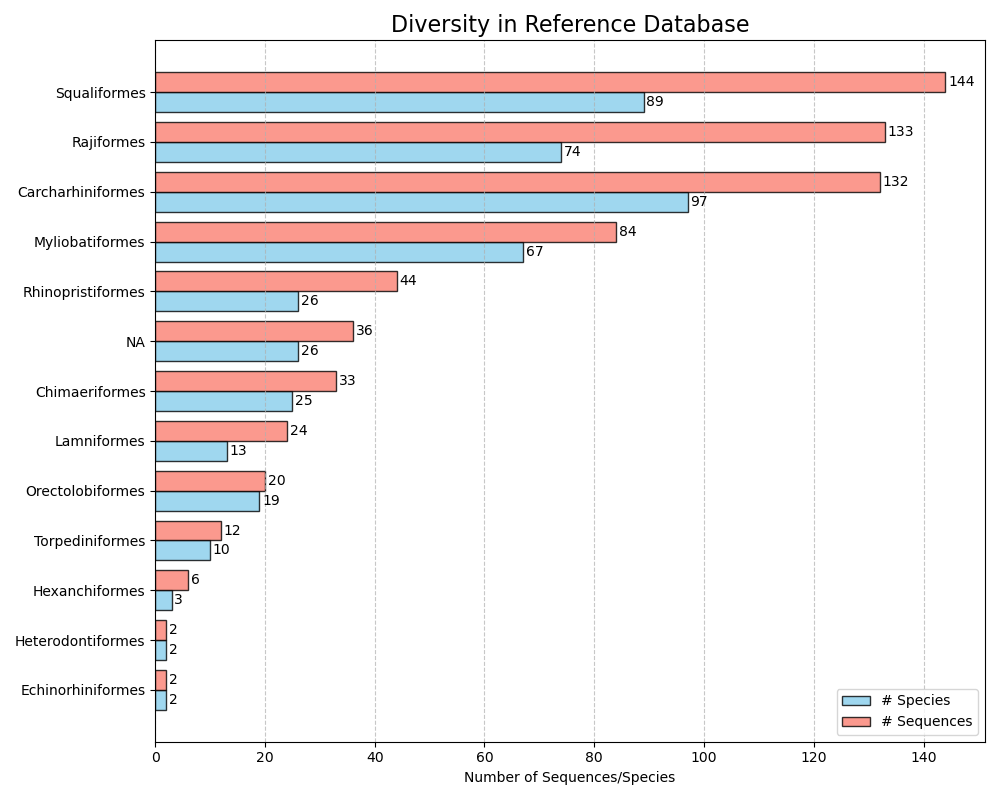
--diversity-figure --diversity-figure関数は、参照データベースの各分類群の種類(青)とシーケンスの数(オレンジ)の数を持つ水平バープロットを生成します。ユーザーは、 --tax-levelパラメーターで参照データベースを分割するために分類ランクを指定できます。税レベルは、 --import関数中に表示されたランクの数です。たとえば、 --ranks 'superkingdom;phylum;class;order;family;genus;species'が使用された場合--importスーパーキングドムに基づいてインパート分割が必要になります--tax-level 1 、phylum = --tax-level 2 、 class = --tax-level 3など。Crabs形式の入力ファイルは、 --inputパラメーターを使用して指定できます。 .png形式の図は、 --outputパラメーターを使用して指定できる出力ファイルに書き込まれます。
crabs --diversity-figure --input crabs_testing/subset.txt --output crabs_testing/diversity-figure.png --tax-level 4
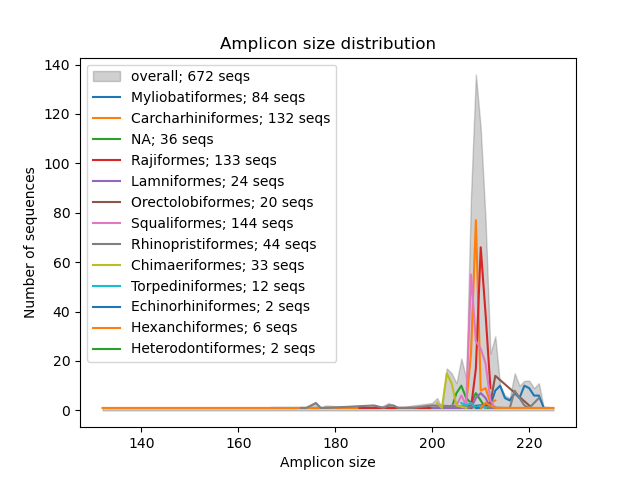
--amplicon-length-figure --amplicon-length-figure関数は、アンプリコン長の範囲を表示する線グラフを生成します。参照データベース内のすべてのシーケンスにわたるアンプリコン長の全体的な範囲は日陰の灰色で表示されますが、結果は分類群(パラメーター: --tax-level )ごとに分割されます。さらに、凡例は、各分類群に割り当てられたシーケンスの数と、参照データベース内のシーケンスの総数を表示します。 CRABS形式の入力ファイルは、 --inputパラメーターを使用して指定できます。 .png形式の図は、 --outputパラメーターを使用して指定できる出力ファイルに書き込まれます。
crabs --amplicon-length-figure --input crabs_testing/subset.txt --output crabs_testing/amplicon-length-figure.png --tax-level 4
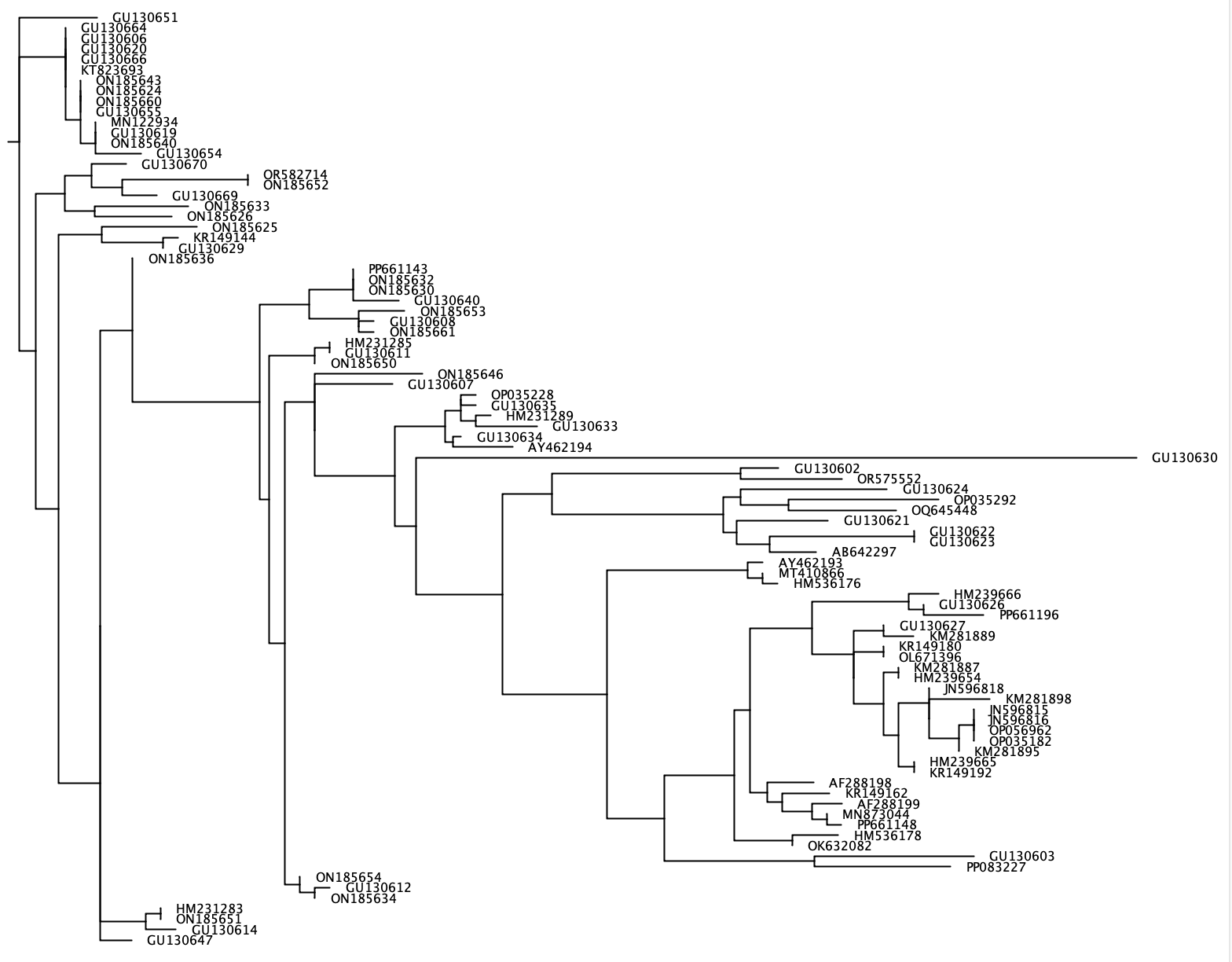
--phylogenetic-tree --phylogenetic-tree関数は、関心のある種のリストのために系統樹を生成します。関心のある種のリストは、 --speciesパラメーターを使用してインポートでき、各ラインに+の種名を持つ.txtファイルで区切られた入力文字列のいずれかで構成されます。関心のある各種について、シーケンスは、ユーザー定義の分類ランク(パラメーター: --tax-level )を関心のある種と共有するリファレンスデータベースから抽出されます。 Crabsは、ClustAlw2 V 2.1を使用して抽出されたすべての配列のアラインメントを生成し、FastTreeを使用して近隣に結合する系統樹を生成します。 Newick形式のこの系統樹は、 --outputパラメーターを使用して出力ファイルに書き込まれ、FigtreeやGeneiousなどのソフトウェアプログラムで視覚化できます。対象の種ごとに別の系統樹が生成されるため、 --output一般的なファイル名を取ります。一方、正確な出力ファイルには、この汎用名が含まれ、その後に「_species_name.tree」が含まれます。
crabs --phylogenetic-tree --input crabs_testing/subset.txt --output crabs_testing/phylo --tax-level 4 --species 'Carcharodon carcharias+Squalus acanthias'
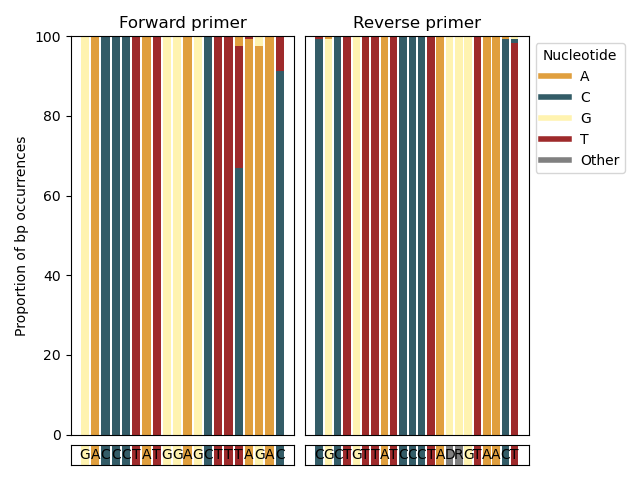
--amplification-efficiency-figure --amplification-efficiency-figure関数は棒グラフを生成し、ユーザー指定の分類群のプライマー結合領域でのベースペアの発生の割合を表示し、それにより、不一致がある前後のプライマー結合領域の場所を視覚化します関心のある分類学的グループで発生している可能性があり、増幅効率に影響を与える可能性があります。 --amplification-efficiency-figure関数は、 --ampliconsパラメーターを使用して入力として最終的なCRABS形式の参照データベースを取得します。入力ファイルの各シーケンスのプライマー結合領域に関する情報を見つけるには、 --inputパラメーターを使用して、インポート後に最初にダウンロードされたシーケンスを提供する必要があります。フォワードおよびリバースプライマーシーケンス(5 '-3'方向)は、 --forwardパラメーターと--reverseパラメーターを使用して提供されます。関心のある分類学的グループの名前は、 --tax-groupパラメーターを使用して提供でき、入力ファイルに組み込まれている分類レベルで設定できます。最後に、.png形式の図は、 --outputパラメーターによって指定された出力ファイルに書き込まれます。
crabs --amplification-efficiency-figure --input crabs_testing/merged.txt --amplicons crabs_testing/subset.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT --output crabs_testing/amplification-efficiency.png --tax-group Carcharhiniformes
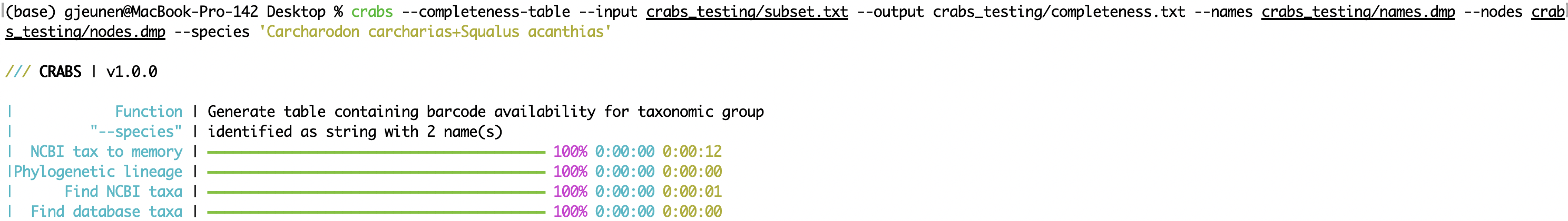
--completeness-table --completeness-table関数は、関心のある種のリストに関する情報を使用して、タブが削除されたテーブル(パラメーター: --output )を出力します。関心のある種のリストは、 --speciesパラメーターを使用してインポートでき、各ラインに+の種名を持つ.txtファイルで区切られた入力文字列のいずれかで構成されます。 「 names.dmp 」および ' nodes.dmp 'ファイルを使用して、それぞれ--namesと--nodesパラメーターを使用して--download-taxonomy 」ファイルを使用して、関心のある種ごとに分類学的系統が生成されます。出力テーブルには、次の情報を提供する10列があります。
crabs --completeness-table --input crabs_testing/subset.txt --output crabs_testing/completeness.txt --names crabs_testing/names.dmp --nodes crabs_testing/nodes.dmp --species 'Carcharodon carcharias+Squalus acanthias'
crabs --version v 1.0.6 :バグ修正 - > --import中の太字のヘッダーの解析の改善。crabs --version v 1.0.5 :バグ修正 - > Blast+ソフトウェアに必要に応じて、BLASTデータベースを構築するときに、SEQ IDに長さの制限を追加しました。crabs --version v 1.0.4 :追加情報 - > --pairwise-global-alignment --coverage --percent-identityの値入力に関する正しい情報を提供しました。crabs --version v 1.0.3 :バグ修正 - >分析を中止する前に3回NCBIサーバーの応答をチェックします。crabs --version v 1.0.2 :バグ修正 - >分析後に0シーケンスが返されたときに報告できます。crabs --version v 1.0.1 :バグ修正 - > --speciesパラメーターを使用したNCBIクエリの構築の成功。

