BindCraft
v1.2.0
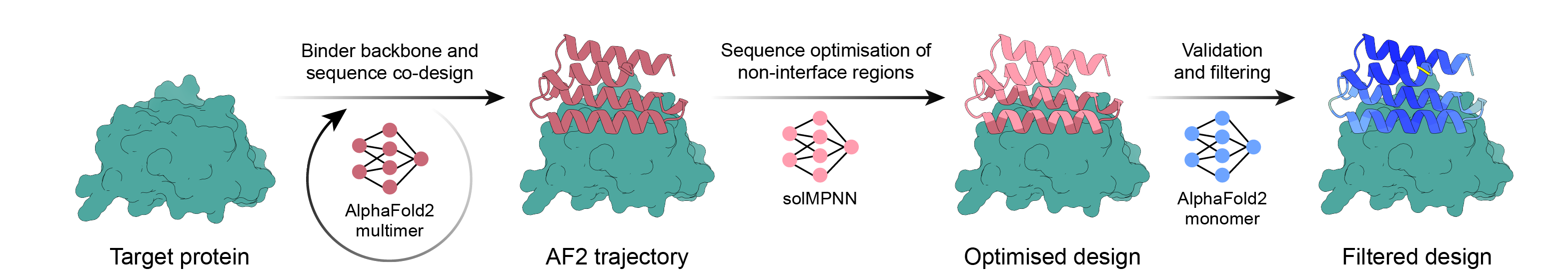
AlphaFold2 バックプロパゲーション、MPNN、および PyRosetta を使用したシンプルなバインダー設計パイプライン。ターゲットを選択し、スクリプトに残りの作業を実行させ、注文する十分なデザインが揃ったら終了します。
BindCraft のプレプリント リンク
まず、このリポジトリのクローンを作成する必要があります。 [install_folder] をインストールするパスに置き換えます。
git clone https://github.com/martinpacesa/BindCraft [install_folder]
cd を使用してインストール フォルダーに移動し、インストール コードを実行します。 BindCraft を実行するには、CUDA 互換の Nvidia グラフィック カードが必要です。 cuda設定では、グラフィックス カードと互換性のある CUDA バージョン (例: 11.8) を指定してください。不明な場合は空白のままにしますが、インストール時に間違ったバージョンが選択され、エラーが発生する可能性があります。 pkg_managerでは、「mamba」と「conda」のどちらを使用しているかを指定します。空白のままにすると、デフォルトで「conda」が使用されます。
注: このインストール スクリプトは PyRosetta をインストールしますが、商用目的の場合はライセンスが必要です。
bash install_bindcraft.sh --cuda '12.4' --pkg_manager 'conda'
常に入力ターゲット PDB を可能な限り小さいサイズにトリミングするようにしてください。これにより、バインダーの生成が大幅に高速化され、GPU メモリ要件が最小限に抑えられます。
受け入れられるバインダーを確認するには、少なくとも数百の軌道を実行する準備をしてください。難しいターゲットの場合は、数千の軌道を実行することもあります。
スクリプトをローカルで実行するには、まずsettings_targetフォルダーでターゲット .json ファイルを構成する必要があります。 json ファイルには次の設定があります。
design_path -> path where to save designs and statistics
binder_name -> what to prefix your designed binder files with
starting_pdb -> the path to the PDB of your target protein
chains -> which chains to target in your protein, rest will be ignored
target_hotspot_residues -> which position to target for binder design, for example `1,2-10` or chain specific `A1-10,B1-20` or entire chains `A`, set to null if you want AF2 to select binding site; better to select multiple target residues or a small patch to reduce search space for binder
lengths -> range of binder lengths to design
number_of_final_designs -> how many designs that pass all filters to aim for, script will stop if this many are reached
次に、バインダー設計スクリプトを実行します。
sbatch ./bindcraft.slurm --settings './settings_target/PDL1.json' --filters './settings_filters/default_filters.json' --advanced './settings_advanced/default_4stage_multimer.json'
設定フラグは、上で設定したターゲット .json を指す必要があります。フィルターフラグは、デザイン フィルターが指定されている json を指します (デフォルトは ./filters/default_filters.json)。詳細フラグは、詳細設定を指します (デフォルトは ./advanced_settings/default_4stage_multimer.json)。フィルターと詳細設定フラグを省略すると、自動的にデフォルトが参照されます。
または、マシンが SLURM をサポートしていない場合は、conda で環境をアクティブ化し、Python コードを実行することで、コードを直接実行できます。
conda activate BindCraft
cd /path/to/bindcraft/folder/
python -u ./bindcraft.py --settings './settings_target/PDL1.json' --filters './settings_filters/default_filters.json' --advanced './settings_advanced/default_4stage_multimer.json'
すべてのフィルターを通過した少なくとも 100 個の最終デザインを生成し、実験的な特性評価のために上位 5 ~ 20 個を注文することをお勧めします。高親和性バインダーが必要な場合、ランク付けに使用される ipTM メトリクスは親和性の良好な予測因子ではありませんが、結合の二値予測因子としては優れていることが示されているため、より多くのスクリーニングを行う方が良いでしょう。
以下に、個々のフィルターと詳細設定について説明します。
設計プロセスを制御する詳細設定は次のとおりです。
omit_AAs -> which amino acids to exclude from design (note: they can still occur if no other options are possible in the position)
force_reject_AA -> whether to force reject design if it contains any amino acids specified in omit_AAs
design_algorithm -> which design algorithm for the trajecory to use, the currently implemented algorithms are below
use_multimer_design -> whether to use AF2-ptm or AF2-multimer for binder design; the other model will be used for validation then
num_recycles_design -> how many recycles of AF2 for design
num_recycles_validation -> how many recycles of AF2 use for structure prediction and validation
sample_models = True -> whether to randomly sample parameters from AF2 models, recommended to avoid overfitting
rm_template_seq_design -> remove target template sequence for design (increases target flexibility)
rm_template_seq_predict -> remove target template sequence for reprediction (increases target flexibility)
rm_template_sc_design -> remove sidechains from target template for design
rm_template_sc_predict -> remove sidechains from target template for reprediction
# Design iterations
soft_iterations -> number of soft iterations (all amino acids considered at all positions)
temporary_iterations -> number of temporary iterations (softmax, most probable amino acids considered at all positions)
hard_iterations -> number of hard iterations (one hot encoding, single amino acids considered at all positions)
greedy_iterations -> number of iterations to sample random mutations from PSSM that reduce loss
greedy_percentage -> What percentage of protein length to mutate during each greedy iteration
# Design weights, higher value puts more weight on optimising the parameter.
weights_plddt -> Design weight - pLDDT of designed chain
weights_pae_intra -> Design weight - PAE within designed chain
weights_pae_inter -> Design weight - PAE between chains
weights_con_intra -> Design weight - maximise number of contacts within designed chain
weights_con_inter -> Design weight - maximise number of contacts between chains
intra_contact_distance -> Cbeta-Cbeta cutoff distance for contacts within the binder
inter_contact_distance -> Cbeta-Cbeta cutoff distance for contacts between binder and target
intra_contact_number -> how many contacts each contact esidue should make within a chain, excluding immediate neighbours
inter_contact_number -> how many contacts each contact residue should make between chains
weights_helicity -> Design weight - helix propensity of the design, Default 0, negative values bias towards beta sheets
random_helicity -> whether to randomly sample helicity weights for trajectories, from -1 to 1
# Additional losses
use_i_ptm_loss -> Use i_ptm loss to optimise for interface pTM score?
weights_iptm -> Design weight - i_ptm between chains
use_rg_loss -> use radius of gyration loss?
weights_rg -> Design weight - radius of gyration weight for binder
use_termini_distance_loss -> Try to minimise distance between N- and C-terminus of binder? Helpful for grafting
weights_termini_loss -> Design weight - N- and C-terminus distance minimisation weight of binder
# MPNN settings
mpnn_fix_interface -> whether to fix the interface designed in the starting trajectory
num_seqs -> number of MPNN generated sequences to sample and predict per binder
max_mpnn_sequences -> how many maximum MPNN sequences per trajectory to save if several pass filters
max_tm-score_filter -> filter out final lower ranking designs by this TM score cut off relative to all passing designs
max_seq-similarity_filter -> filter out final lower ranking designs by this sequence similarity cut off relative to all passing designs
sampling_temp = 0.1 -> sampling temperature for amino acids, T=0.0 means taking argmax, T>>1.0 means sampling randomly.")
# MPNN settings - advanced
sample_seq_parallel -> how many sequences to sample in parallel, reduce if running out of memory
backbone_noise -> backbone noise during sampling, 0.00-0.02 are good values
model_path -> path to the MPNN model weights
mpnn_weights -> whether to use "original" mpnn weights or "soluble" weights
save_mpnn_fasta -> whether to save MPNN sequences as fasta files, normally not needed as the sequence is also in the CSV file
# AF2 design settings - advanced
num_recycles_design -> how many recycles of AF2 for design
num_recycles_validation -> how many recycles of AF2 use for structure prediction and validation
optimise_beta -> optimise predictions if beta sheeted trajectory detected?
optimise_beta_extra_soft -> how many extra soft iterations to add if beta sheets detected
optimise_beta_extra_temp -> how many extra temporary iterations to add if beta sheets detected
optimise_beta_recycles_design -> how many recycles to do during design if beta sheets detected
optimise_beta_recycles_valid -> how many recycles to do during reprediction if beta sheets detected
# Optimise script
remove_unrelaxed_trajectory -> remove the PDB files of unrelaxed designed trajectories, relaxed PDBs are retained
remove_unrelaxed_complex -> remove the PDB files of unrelaxed predicted MPNN-optimised complexes, relaxed PDBs are retained
remove_binder_monomer -> remove the PDB files of predicted binder monomers after scoring to save space
zip_animations -> at the end, zip Animations trajectory folder to save space
zip_plots -> at the end, zip Plots trajectory folder to save space
save_trajectory_pickle -> save pickle file of the generated trajectory, careful, takes up a lot of storage space!
max_trajectories -> how many maximum trajectories to generate, for benchmarking
acceptance_rate -> what fraction of trajectories should yield designs passing the filters, if the proportion of successful designs is less than this fraction then the script will stop and you should adjust your design weights
start_monitoring -> after what number of trajectories should we start monitoring acceptance_rate, do not set too low, could terminate prematurely
# debug settings
enable_mpnn = True -> whether to enable MPNN design
enable_rejection_check -> enable rejection rate check
デザインをフィルタリングするための機能は次のとおりです。一部を使用したくない場合は、しきい値としてnullを設定するだけです。より高い値は、しきい値より高い値を維持するか (true)、低い値を維持するか (false) を示します。 N_ で始まる特徴は、各 AlphaFold モデルごとの統計に対応します。平均は予測されたすべてのモデルにわたるものです。
MPNN_score -> MPNN sequence score, generally not recommended as it depends on protein
MPNN_seq_recovery -> MPNN sequence recovery of original trajectory
pLDDT -> pLDDT confidence score of AF2 complex prediction, normalised to 0-1
pTM -> pTM confidence score of AF2 complex prediction, normalised to 0-1
i_pTM -> interface pTM confidence score of AF2 complex prediction, normalised to 0-1
pAE -> predicted alignment error of AF2 complex prediction, normalised compared AF2 by n/31 to 0-1
i_pAE -> predicted interface alignment error of AF2 complex prediction, normalised compared AF2 by n/31 to 0-1
i_pLDDT -> interface pLDDT confidence score of AF2 complex prediction, normalised to 0-1
ss_pLDDT -> secondary structure pLDDT confidence score of AF2 complex prediction, normalised to 0-1
Unrelaxed_Clashes -> number of interface clashes before relaxation
Relaxed_Clashes -> number of interface clashes after relaxation
Binder_Energy_Score -> Rosetta energy score for binder alone
Surface_Hydrophobicity -> surface hydrophobicity fraction for binder
ShapeComplementarity -> interface shape complementarity
PackStat -> interface packstat rosetta score
dG -> interface rosetta dG energy
dSASA -> interface delta SASA (size)
dG/dSASA -> interface energy divided by interface size
Interface_SASA_% -> Fraction of binder surface covered by the interface
Interface_Hydrophobicity -> Interface hydrophobicity fraction of binder interface
n_InterfaceResidues -> number of interface residues
n_InterfaceHbonds -> number of hydrogen bonds at the interface
InterfaceHbondsPercentage -> number of hydrogen bonds compared to interface size
n_InterfaceUnsatHbonds -> number of unsatisfied buried hydrogen bonds at the interface
InterfaceUnsatHbondsPercentage -> number of unsatisfied buried hydrogen bonds compared to interface size
Interface_Helix% -> proportion of alfa helices at the interface
Interface_BetaSheet% -> proportion of beta sheets at the interface
Interface_Loop% -> proportion of loops at the interface
Binder_Helix% -> proportion of alfa helices in the binder structure
Binder_BetaSheet% -> proportion of beta sheets in the binder structure
Binder_Loop% -> proportion of loops in the binder structure
InterfaceAAs -> number of amino acids of each type at the interface
HotspotRMSD -> unaligned RMSD of binder compared to original trajectory, in other words how far is binder in the repredicted complex from the original binding site
Target_RMSD -> RMSD of target predicted in context of the designed binder compared to input PDB
Binder_pLDDT -> pLDDT confidence score of binder predicted alone
Binder_pTM -> pTM confidence score of binder predicted alone
Binder_pAE -> predicted alignment error of binder predicted alone
Binder_RMSD -> RMSD of binder predicted alone compared to original trajectory
コーディングとアイデアの議論に協力してくれた Lennart Nickel、Yehlin Cho、Casper Goverde、Sergey Ovchinnikov に感謝します。このリポジトリは、次のコードを使用します。


