nucleotide transformer
1.0.0
この InstaDeep Github リポジトリへようこそ。以下の内容が紹介されています。
私たちはこれらの作品をオープンソース化し、これら 9 つのゲノミクス言語モデルと 2 つのセグメンテーション モデルのコードと事前トレーニングされた重みへのアクセスをコミュニティに提供できることを嬉しく思います。 nucleotide transformerプロジェクトのモデルは Nvidia および TUM と協力して開発され、モデルは Cambridge-1 の DGX A100 ノードでトレーニングされました。 Agro nucleotide transformerプロジェクトのモデルは Google と共同で開発され、モデルは TPU-v4 アクセラレーターでトレーニングされました。
全体として、私たちの研究は、言語基礎モデルの事前トレーニングと適用、およびそれらをバックボーンエンコーダーとして使用するモデルのトレーニングに関連する新しい洞察をゲノミクスに提供し、現場での応用の十分な機会を提供します。
このリポジトリには次のものが含まれます。
他のアプローチと比較して、当社のモデルは単一の参照ゲノムからの情報を統合するだけでなく、3,200 を超える多様なヒトゲノム、およびモデル生物と非モデル生物を含む広範囲の種からの 850 のゲノムからの DNA 配列を活用します。堅牢かつ広範な評価を通じて、これらの大規模モデルが既存の方法と比較して非常に正確な分子表現型予測を提供することを示します。
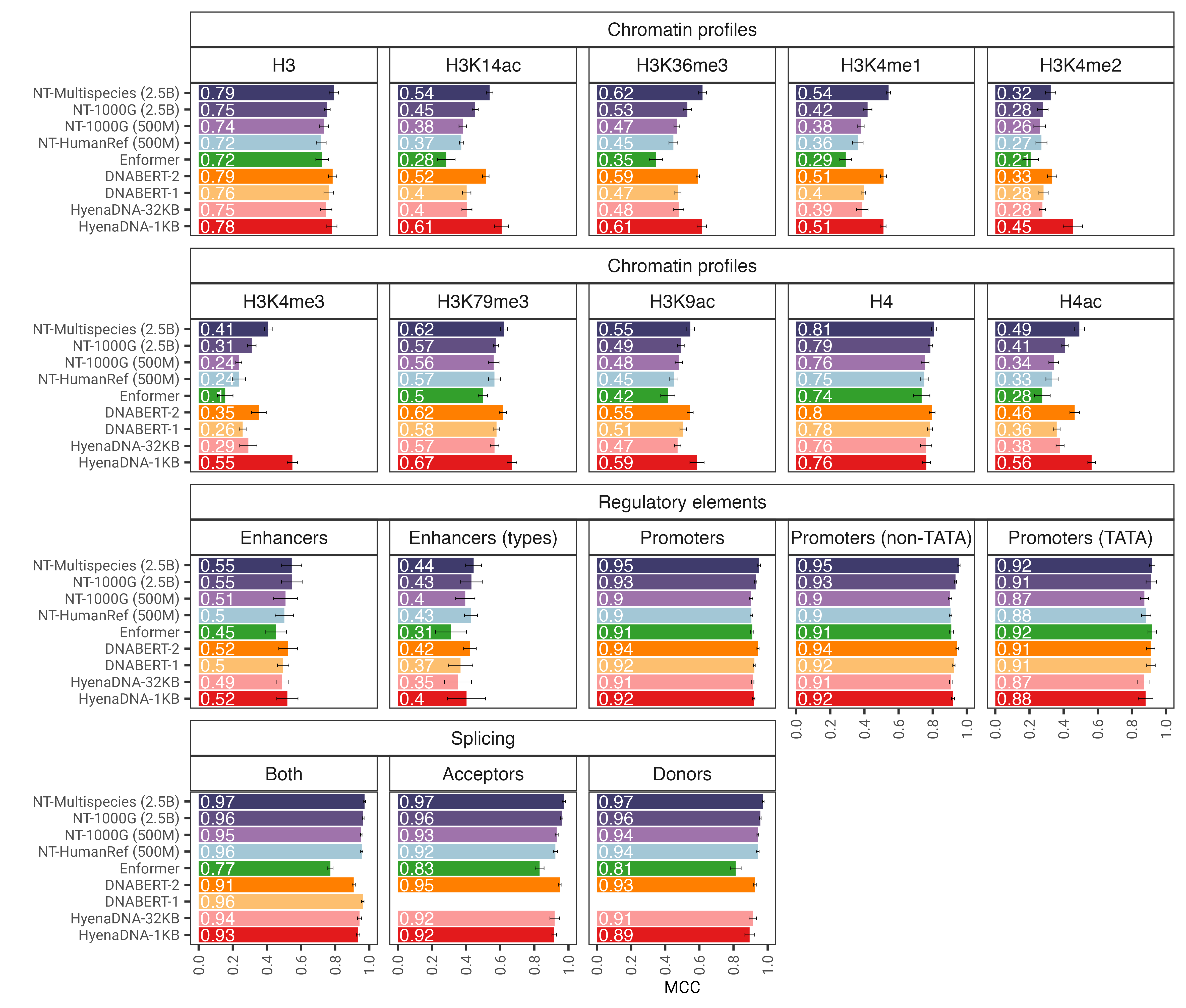
図 1: nucleotide transformerモデルは、微調整後にさまざまなゲノミクスタスクを正確に予測します。微調整された変圧器モデルの下流タスク全体のパフォーマンス結果を示します。エラーバーは、10 分割交差検証から得られた 2 つの SD を表します。
この研究では、主に作物種に焦点を当てた 48 植物種の参照ゲノムでトレーニングされた、新しい基礎的な大規模言語モデルを紹介します。私たちは、制御機能、RNA プロセシング、遺伝子発現に至るまでのいくつかの予測タスクにわたって AgroNT のパフォーマンスを評価し、AgroNT が最先端のパフォーマンスを獲得できることを示しました。
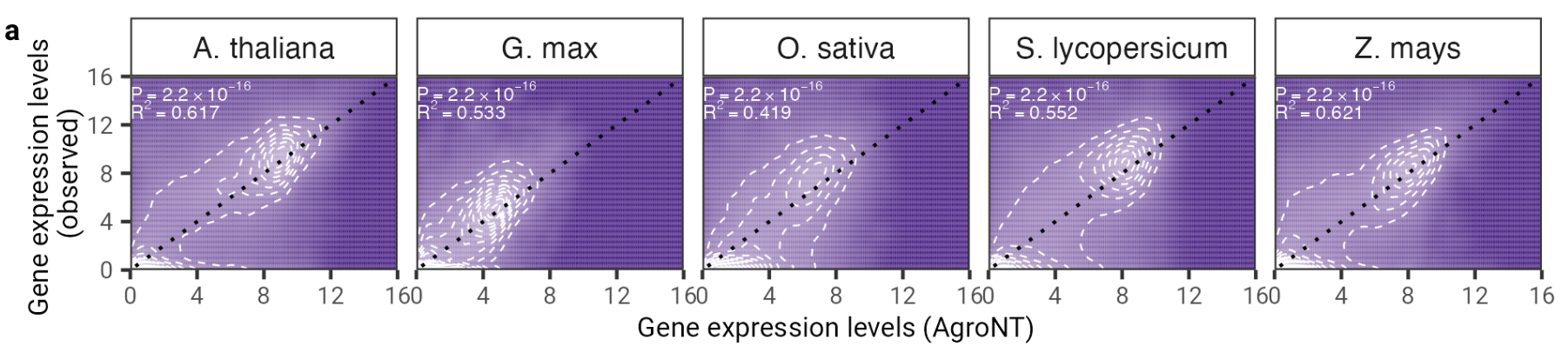
図 2: AgroNT は、さまざまな植物種にわたる遺伝子発現予測を提供します。すべての組織にわたるホールドアウト遺伝子の遺伝子発現予測は、観察された遺伝子発現レベルと相関しています。線形モデルからの決定係数 (R 2 ) と、予測値と観測値の間の関連する P 値が示されています。
コードと事前トレーニングされたモデルを使用するには、次のようにします。
pip install . 。その後、ダウンロードして、わずか数行のコードで 9 つのモデルのいずれかを使用して推論を実行できます。
import haiku as hk
import jax
import jax . numpy as jnp
from nucleotide_transformer . pretrained import get_pretrained_model
# Get pretrained model
parameters , forward_fn , tokenizer , config = get_pretrained_model (
model_name = "500M_human_ref" ,
embeddings_layers_to_save = ( 20 ,),
max_positions = 32 ,
)
forward_fn = hk . transform ( forward_fn )
# Get data and tokenize it
sequences = [ "ATTCCGATTCCGATTCCG" , "ATTTCTCTCTCTCTCTGAGATCGATCGATCGAT" ]
tokens_ids = [ b [ 1 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens_str = [ b [ 0 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens = jnp . asarray ( tokens_ids , dtype = jnp . int32 )
# Initialize random key
random_key = jax . random . PRNGKey ( 0 )
# Infer
outs = forward_fn . apply ( parameters , random_key , tokens )
# Get embeddings at layer 20
print ( outs [ "embeddings_20" ]. shape )サポートされているモデル名は次のとおりです。
Google Colab でモデルを実行し、その他のサンプル コードを見つけることもできます。
Jax のおかげで、コードは GPU と TPU の両方で実行されます。
私たちの 2 番目のバージョンのnucleotide transformer v2 モデルには、より効率的であることが証明された一連のアーキテクチャ上の変更が含まれています。学習された位置埋め込みを使用する代わりに、各アテンション レイヤーで使用されるロータリー 埋め込みと、バイアスのないスウィッシュ アクティベーションを備えたゲート線形ユニットを使用します。これらの改良されたモデルは、最大 2,048 個のトークンのシーケンスも受け入れ、コンテキスト ウィンドウが 12kbp と長くなります。チンチラのスケーリング則にヒントを得て、v1 モデル (300B トークン) と比較して、複数種のデータセットで NT-v2 モデルを長時間トレーニングしました (50M および 100M モデルの場合は 300B トークン、250M および 500M モデルの場合は 1T トークン)。 4 つのモデルすべて)。
トランスフォーマー層には 1 のインデックスが付いています。これは、引数model_name="500M_human_ref"およびembeddings_layers_to_save=(1, 20,)を指定してget_pretrained_model呼び出すと、最初と 20 番目のトランスフォーマー層の後に埋め込みが抽出されることを意味します。 Roberta LM ヘッドを使用する変圧器の場合、最後の変圧器ブロックの後ではなく、LM ヘッドの最初の層ノルムの後に最終的な埋め込みを抽出するのが一般的です。したがって、 get_pretrained_model次の引数embeddings_layers_to_save=(24,)を指定して呼び出された場合、埋め込みは最後の変換層の後で抽出されるのではなく、LM ヘッドの最初の層ノルムの後に抽出されます。
SegmentNT モデルは、言語モデル ヘッドを削除し、1 次元 U-Net セグメンテーション ヘッドに置き換えたnucleotide transformer (NT) トランスフォーマーを利用して、単一ヌクレオチド解像度で配列内の数種類のゲノミクス要素の位置を予測します。最大 30 kb の入力配列内の 14 の異なるクラスのヒトゲノミクス要素に関する 2 つの異なるモデル バリアントを提示します。これらには、遺伝子 (タンパク質コード遺伝子、lncRNA、5'UTR、3'UTR、エクソン、イントロン、スプライスアクセプターおよびドナー部位) および調節 (ポリA シグナル、組織不変および組織特異的プロモーターおよびエンハンサー、CTCF 結合) が含まれます。サイト)要素。 SegmentNT は、NT の事前トレーニングされた重みの恩恵を受けて、最先端の U-Net セグメンテーション アーキテクチャよりも優れたパフォーマンスを実現し、最大 50kbp のゼロショット汎化を実証します。
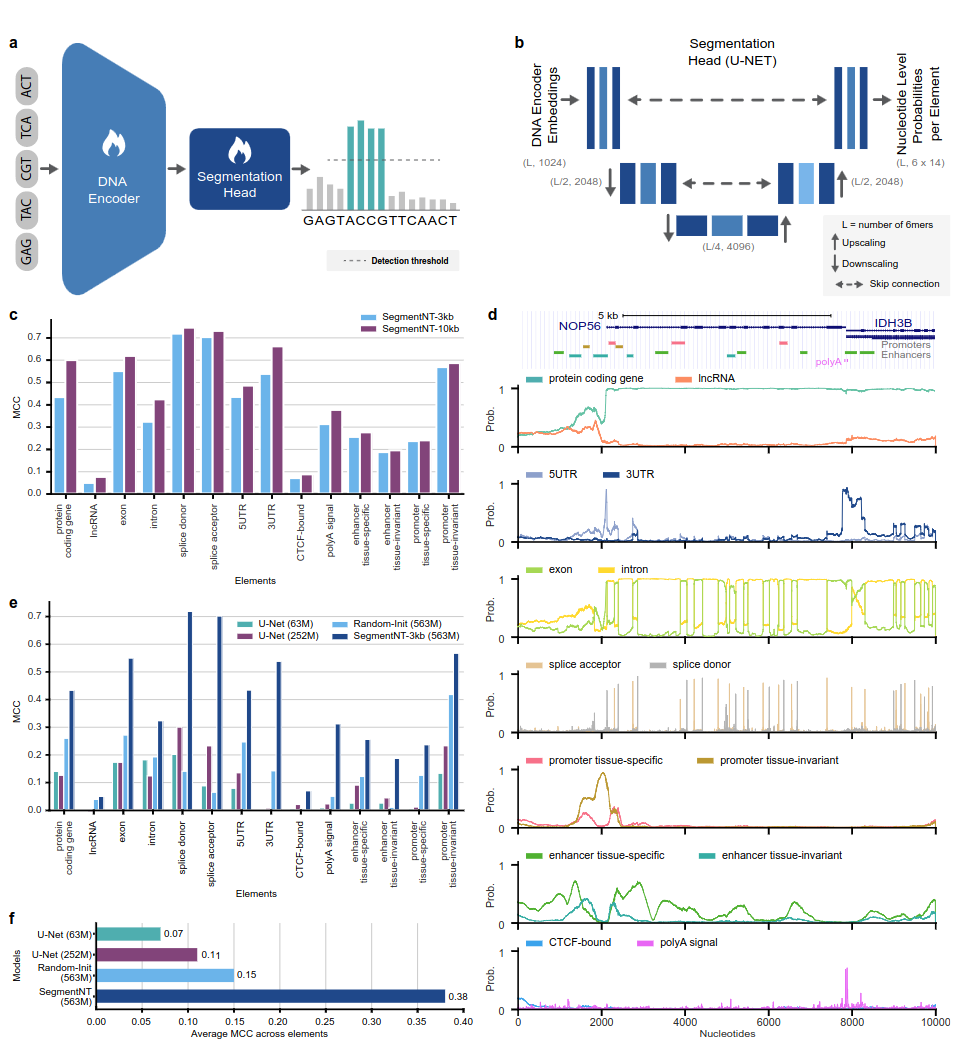
図 1: SegmentNT は、ヌクレオチド解像度でゲノミクス要素の位置を特定します。
コードと事前トレーニングされたモデルを使用するには、次のようにします。
pip install . 。その後、ダウンロードして、わずか数行のコードで任意のモデルを使用してシーケンスを推論できます。
rescaling factorトレーニング中に使用された係数に設定されます。 30kbp と 50kbp の間のシーケンスを推論する必要がある場合は、 get_pretrained_segment_nt_model関数のrescaling_factor引数に、値rescaling_factor = max_num_nucleotides / max_num_tokens_ntを指定して渡します。ここで、 num_dna_tokens_inferenceは推論時のトークンの数です (つまり、40008 のシーケンスの場合は 6669)。 max_num_tokens_ntは、バックボーン ヌクレオチド トランスフォーマーがトレーニングされたトークンの最大数、つまり2048です。
?ノートブックのexamples/inference_segment_nt.ipynbでは、50 kb シーケンスを推論し、論文の図 3 を再現する確率をプロットする方法が示されています。
? SegmentNT モデルは、各ヌクレオチドを 6 量体としてトークン化する必要があるため、入力配列内の「N」を処理しません。これは、1 つまたは複数の「N」塩基対を含む配列を使用する場合には当てはまりません。
import haiku as hk
import jax
import jax . numpy as jnp
from nucleotide_transformer . pretrained import get_pretrained_segment_nt_model
# Initialize CPU as default JAX device. This makes the code robust to memory leakage on
# the devices.
jax . config . update ( "jax_platform_name" , "cpu" )
backend = "cpu"
devices = jax . devices ( backend )
num_devices = len ( devices )
print ( f"Devices found: { devices } " )
# The number of DNA tokens (excluding the CLS token prepended) needs to be dividible by
# 2 to the power of the number of downsampling block, i.e 4.
max_num_nucleotides = 8
assert max_num_nucleotides % 4 == 0 , (
"The number of DNA tokens (excluding the CLS token prepended) needs to be dividible by"
"2 to the power of the number of downsampling block, i.e 4." )
parameters , forward_fn , tokenizer , config = get_pretrained_segment_nt_model (
model_name = "segment_nt" ,
embeddings_layers_to_save = ( 29 ,),
attention_maps_to_save = (( 1 , 4 ), ( 7 , 10 )),
max_positions = max_num_nucleotides + 1 ,
)
forward_fn = hk . transform ( forward_fn )
apply_fn = jax . pmap ( forward_fn . apply , devices = devices , donate_argnums = ( 0 ,))
# Get data and tokenize it
sequences = [ "ATTCCGATTCCGATTCCAACGGATTATTCCGATTAACCGATTCCAATT" , "ATTTCTCTCTCTCTCTGAGATCGATGATTTCTCTCTCATCGAACTATG" ]
tokens_ids = [ b [ 1 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens_str = [ b [ 0 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens = jnp . asarray ( tokens_ids , dtype = jnp . int32 )
random_key = jax . random . PRNGKey ( seed = 0 )
keys = jax . device_put_replicated ( random_key , devices = devices )
parameters = jax . device_put_replicated ( parameters , devices = devices )
tokens = jax . device_put_replicated ( tokens , devices = devices )
# Infer on the sequence
outs = apply_fn ( parameters , keys , tokens )
# Obtain the logits over the genomic features
logits = outs [ "logits" ]
# Transform them in probabilities
probabilities = jnp . asarray ( jax . nn . softmax ( logits , axis = - 1 ))[..., - 1 ]
print ( f"Probabilities shape: { probabilities . shape } " )
print ( f"Features inferred: { config . features } " )
# Get probabilities associated with intron
idx_intron = config . features . index ( "intron" )
probabilities_intron = probabilities [..., idx_intron ]
print ( f"Intron probabilities shape: { probabilities_intron . shape } " )サポートされているモデル名は次のとおりです。
Jax のおかげで、コードは GPU と TPU の両方で実行されます。
モデルは、シーケンスの先頭に自動的に付加される <CLS> トークンを含む、最大 1000 トークンの長さのシーケンスでトレーニングされます。トークナイザーは、文字「A」、「C」、「G」、「T」を 6 マーにグループ化することで、左から右にトークナイズを開始します。 「N」文字は、k-mer 内でグループ化されないように選択されているため、トークナイザーが「N」に遭遇するたび、または配列内のヌクレオチドの数が 6 の倍数でない場合は、グループ化せずにヌクレオチドをトークン化します。彼ら。以下に例を示します。
dna_sequence_1 = "ACGTGTACGTGCACGGACGACTAGTCAGCA"
tokenized_dna_sequence_1 = [ < CLS > , < ACGTGT > , < ACGTGC > , < ACGGAC > , < GACTAG > , < TCAGCA > ]
dna_sequence_2 = "ACGTGTACNTGCACGGANCGACTAGTCTGA"
tokenized_dna_sequence_2 = [ < CLS > , < ACGTGT > , < A > , < C > , < N > , < TGCACG > , < G > , < A > , < N > , < CGACTA > , < GTCTGA > ]したがって、すべての v1 および v2 トランスフォーマーは、内部に「N」がない場合、それぞれ最大 5994 ヌクレオチドおよび 12282 ヌクレオチドの配列を取ることができます。
このリポジトリで紹介されているモデルのコレクションは、Instadeep の ハグフェイス スペースnucleotide transformerのスペースとアグロnucleotide transformerスペース) で利用できます。
我々は、興味深い研究の方向性を特定するのに役立つ建設的な議論をしてくれたMaša Roller氏、そしてRostlabのメンバー、特にTobias Olenyi氏、Ivan Koludarov氏、Burkhard Rost氏に感謝します。さらに、実験データを公開データベースに保管しているすべての人々、これらのデータベースを維持している人々、および分析および予測方法を自由に利用できるようにしているすべての人々に感謝の意を表します。 Jax 開発チームにも感謝します。
このリポジトリがあなたの仕事に役立つと思われる場合は、関連する論文のいずれかに関連する引用を追加してください。
nucleotide transformerの論文:
@article { dalla2023nucleotide ,
title = { The nucleotide transformer : Building and Evaluating Robust Foundation Models for Human Genomics } ,
author = { Dalla-Torre, Hugo and Gonzalez, Liam and Mendoza Revilla, Javier and Lopez Carranza, Nicolas and Henryk Grywaczewski, Adam and Oteri, Francesco and Dallago, Christian and Trop, Evan and Sirelkhatim, Hassan and Richard, Guillaume and others } ,
journal = { bioRxiv } ,
pages = { 2023--01 } ,
year = { 2023 } ,
publisher = { Cold Spring Harbor Laboratory }
}農業用nucleotide transformer紙:
@article { mendoza2024foundational ,
title = { A foundational large language model for edible plant genomes } ,
author = { Mendoza-Revilla, Javier and Trop, Evan and Gonzalez, Liam and Roller, Ma{v{s}}a and Dalla-Torre, Hugo and de Almeida, Bernardo P and Richard, Guillaume and Caton, Jonathan and Lopez Carranza, Nicolas and Skwark, Marcin and others } ,
journal = { Communications Biology } ,
volume = { 7 } ,
number = { 1 } ,
pages = { 835 } ,
year = { 2024 } ,
publisher = { Nature Publishing Group UK London }
}セグメントNT紙
@article { de2024segmentnt ,
title = { SegmentNT: annotating the genome at single-nucleotide resolution with DNA foundation models } ,
author = { de Almeida, Bernardo P and Dalla-Torre, Hugo and Richard, Guillaume and Blum, Christopher and Hexemer, Lorenz and Gelard, Maxence and Pandey, Priyanka and Laurent, Stefan and Laterre, Alexandre and Lang, Maren and others } ,
journal = { bioRxiv } ,
pages = { 2024--03 } ,
year = { 2024 } ,
publisher = { Cold Spring Harbor Laboratory }
}コードやモデルに関するご質問やフィードバックがございましたら、お気軽にお問い合わせください。
私たちの仕事にご興味をお持ちいただきありがとうございます。


