BRAKER
v3.0.8
Breakerに関する最初のBGA23ワークショップセッションの録音です。ビデオを見ることで学習する場合は、それを見ることを検討してください:https://www.youtube.com/watch?v=uxtkj4mukyg
Braker3は現在https://usegalaxy.eu/にあります
Tsebra&Braker3関連:
Breaker&Augustus関連:
ジェネマーク関連:
マーク・ボロドフスキー、ジョージア工科大学、米国、[email protected]
Tomas Bruna、米国共同ゲノム研究所、[email protected]
Alexandre Lomsazde、Georgia Tech、USA、[email protected]
[A]グレイフズ大学大学、数学およびコンピューターサイエンス研究所、Walther-Rathenau-Str。 47、17489 Greifswald、ドイツ
[b]グレイフスワルド大学、微生物の機能ゲノミクスセンター、Felix-Hausdorff-Str。 8、17489 Greifswald、ドイツ
[C]ジョージア工科大学とエモリー大学ウォレスHコールター生物医学工学部、30332アトランタ、米国
[D] Computational Science and Engineeringの学校、30332アトランタ、米国
[e]モスクワ物理学研究所、モスクワ地域141701、ドルゴプルドニー、ロシア
![braker2-team-2 [fig10]](https://images.downcodes.com/uploads/20250214/img_67aee79a0cb7530.png)
![braker2-team-1 [fig11]](https://images.downcodes.com/uploads/20250214/img_67aee79a0d1eb31.png)
![braker2-team-3 [fig12]](https://images.downcodes.com/uploads/20250214/img_67aee79a0da9c32.png)
![braker2-team-4 [fig13]](https://images.downcodes.com/uploads/20250214/img_67aee79a0e49933.png)
図1:現在のブレイカーの著者、左から右へ:マリオ・スタンケ、アレクサンドル・ロンサゼ、キャサリーナ・J・ホフ、トマス・ブルーナ、ラース・ガブリエル、マーク・ボロドフスキー。科学者のより大きなコミュニティがブレーカーコードに貢献したことを認めます(例:プルリクエストを介して)。
Braker1、Braker2、およびBraker3の開発は、国立衛生研究所(NIH)[GM128145からMBおよびMS]によってサポートされていました。 BRAKER3の開発は、ドイツのメクレンブルクヴォルポンポンポンポンポンポンンの政府によってKJHとMSに付与されたプロジェクトデータコンピテンシーによって部分的に資金提供されていました。
Braker(TSEBRA)のトランスクリプトセレクターは、https://github.com/gaius-augustus/tsebraで入手できます。
Brakerのコアの遺伝子ファインダーの1つであるGeneMark-ETPは、https://github.com/gatech-genemark/genemark-etpで入手できます。
Breakerのコアにある2番目の遺伝子ファインダーであるAugustusは、https://github.com/gaius-augustus/augustusで入手できます。
MiniprotまたはGenomethreaderを使用してトレーニング遺伝子を生成するためのブレーカーパイプラインスピンオフであるGalbaは、https://github.com/gius-augustus/galbaで入手できます。
急速に増加しているシーケンスゲノムの数には、正確な遺伝子構造注釈のために完全に自動化された方法が必要です。この目標を念頭に置いて、Genemark-ET R2とAugustus R3、 R4の組み合わせであるBraker1 R1 R0を開発しました。
ただし、新しいゲノムに注釈を付けるために利用可能なRNA-seqデータの品質は可変であり、場合によってはRNA-seqデータはまったく利用できません。
Braker2は、遺伝子予測ツールの完全に自動化されたトレーニングを可能にするBraker1の拡張です。 RNA-seqおよびタンパク質相同性情報からの外因性証拠は、予測に。
タンパク質相同性情報に依存する他の利用可能な方法とは対照的に、Braker2は、非常に密接に関連する種の注釈がなくても、RNA-seqデータがない場合でも、高い遺伝子予測の精度に達します。
Braker3は、Breaker Suiteの最新のパイプラインです。これにより、完全に自動化されたパイプラインでRNA-seqとタンパク質データの使用が可能になり、Genemark-ETPとAuguartusで非常に信頼性の高い遺伝子を訓練および予測できます。パイプラインの結果は、両方の遺伝子予測ツールの組み合わせた遺伝子セットであり、外因性の証拠から非常に高いサポートを持つ遺伝子のみを含んでいます。
このユーザーガイドでは、同じスクリプト( braker.pl )で実行されるため、Braker1、Braker2、およびBraker3を単にBrakerと呼びます。
高品質のゲノムアセンブリを使用します。ゲノムアセンブリに非常に短い足場が膨大な数がある場合、これらの短い足場はランタイムが劇的に増加する可能性がありますが、予測の精度は向上しません。
ゲノムファイルで単純な足場名を使用します(eg >contig1 >contig1my custom species namesome putative function /more/information/ and lots of special characters %&!*(){} 。 Alignmentプログラムを実行する前に、すべてのFASTAファイルの足場名をシンプルにします。
新しいゲノムで遺伝子を正確に予測するために、ゲノムは繰り返しのためにマスクされるべきです。これにより、反復領域と低い複雑な領域における偽陽性遺伝子構造の予測が回避されます。また、RNA-seqデータをいくつかのツールを使用してゲノムにマッピングするためには、リピートマスキングも不可欠です(Hisat2などの他のRNA-Seqマッパーは、マスキング情報を無視します)。 Genemark-ES/ET/EP/ETPおよびAUGUSTUSの場合、ソフトマスキング(つまり、繰り返し領域を小文字に、他のすべての領域を上級文字に入れます)は、ハードマスキングよりも良い結果につながります(つまり、文字で繰り返し領域の文字を置き換える未知のヌクレオチドのN )。
多くのゲノムには、GeneMark-ES/ET/EP/ETPおよびBRAKER内のアウグストゥスの標準パラメーターで正確に予測される遺伝子構造があります。ただし、一部のゲノムには、クレード固有の特徴、つまり真菌の特別な分岐点モデル、または非標準的なスプライスサイトパターンがあります。オプションセクション[オプション]をお読みください。カスタムオプションのいずれかが標的種のゲノムの遺伝子予測の精度を改善できるかどうかを判断してください。
さらに使用する前に、常に遺伝子予測の結果を確認してください!外因性証拠データを使用して、遺伝子モデルの目視検査にゲノムブラウザーを使用できます。 Brakerは、この目的のためにMakeHubを使用してUCSC Genome Browserのトラックデータハブの生成をサポートしています。
Brakerは主に、Genemark-ES/ET/ETP [F1]のサポートされているトレーニングをサポートするサポートされているアウグストゥスの次のトレーニングを最終的な証拠の統合とサポートするトレーニングをサポートする、主に半拡散した外因性の証拠データ(RNA-seqおよび/またはタンパク質スプライスされたアライメント情報)を特徴としています。遺伝子予測ステップ。ただし、ブレーカーには多くの追加のパイプラインが含まれています。以下では、可能な入力ファイルとパイプラインの概要を示します。
![braker2-main-a [fig1]](https://images.downcodes.com/uploads/20250214/img_67aee79a0eaf534.png)
図2:Breaker Pipeline A:ゲノムデータに関するGeneMark-ESのトレーニングのみ。 ab initio遺伝子予測を伴うaugustus
![braker2-main-b [fig2]](https://images.downcodes.com/uploads/20250214/img_67aee79a0f13f35.png)
図3:Braker Pipeline B:RNA-seqスプライスされたアライメント情報でサポートされているGeneMark-ETのトレーニング、同じスプライスされたアライメント情報を使用したAugustusとの予測。
![braker2-main-c [fig3]](https://images.downcodes.com/uploads/20250214/img_67aee79a0fa1036.png)
図4:ブレーカーパイプラインC:タンパク質スプライスされたアライメント、開始および停止情報のトレーニングGeneMark-EP+、同じ情報を使用したAugustusとの予測に加えて、CDSPARTヒント。ここで使用されるタンパク質は、標的生物までのあらゆる進化距離のものです。
![braker3-main-a [fig4]](https://images.downcodes.com/uploads/20250214/img_67aee79a1010b37.png)
図5:Braker Pipeline D:必要に応じて、ターゲット種のRNA-seqセットのダウンロードとアラインメント。 RNA-seqアラインメントと大規模なタンパク質データベース(タンパク質は進化距離の可能性がある)でサポートされているGeneMark-ETPのトレーニング。その後、Augustusのトレーニングと予測と同じ外因性情報をGeneMark-ETPの結果とともに使用します。最終的な予測は、アウグストゥスとGeneMark-ETPの結果のTSEBRAの組み合わせです。
Braker3の「手動」インストールとそのすべての依存関係は、ルート許可なしに退屈で本当に挑戦的であることを認識しています。したがって、特異点で実行されるように開発されたDockerコンテナを提供します。このコンテナに関するすべての情報は、https://hub.docker.com/r/teambraker/braker3にあります。
要するに、次のように作成します。
singularity build braker3.sif docker://teambraker/braker3:latest
実行:
singularity exec braker3.sif braker.pl
テスト:
singularity exec -B $PWD:$PWD braker3.sif cp /opt/BRAKER/example/singularity-tests/test1.sh .
singularity exec -B $PWD:$PWD braker3.sif cp /opt/BRAKER/example/singularity-tests/test2.sh .
singularity exec -B $PWD:$PWD braker3.sif cp /opt/BRAKER/example/singularity-tests/test3.sh .
export BRAKER_SIF=/your/path/to/braker3.sif # may need to modify
bash test1.sh
bash test2.sh
bash test3.sh
Docker内で分析を実行したいユーザーはほとんどいません(ルート許可が必要であるため)。ただし、それがあなたの目標である場合、次のようにコンテナを実行してテストすることができます
sudo docker run --user 1000:100 --rm -it teambraker/braker3:latest bash
bash /opt/BRAKER/example/docker-tests/test1.sh # BRAKER1
bash /opt/BRAKER/example/docker-tests/test2.sh # BRAKER2
bash /opt/BRAKER/example/docker-tests/test3.sh # BRAKER3
幸運を ;-)
$PATH変数に残る古いGeneMarkバージョンは、予期せぬ干渉につながり、プログラムの障害を引き起こす可能性があります。すべての古いGenemarkバージョンを$PATH ( ProtHint/dependenciesのGenemarkなど)から移動してください。
リリース時に、このBreakerバージョンは以下でテストされました。
Augustus 3.5.0 F2
genemark-etp(ソースDockerfileを参照)
Bamtools 2.5.1 R5
Samtools 1.7-4-G93586ed R6
Spaln 2.3.3d R8、 R9、 R10
NCBI BLAST+ 2.2.31+ R12、 R13
ダイヤモンド0.9.24
CDBFASTA 0.99
CDBYANK 0.981
Gushr 1.0.0
SRA Toolkit 3.00 R14
HISAT2 2.2.1 R15
Bedtools 2.30 R16
Stringtie2 2.2.1 R17
GFFREAD 0.12.7 R18
CompleaSm 0.2.5 R27
Breakerを実行するには、 bashとPerlを備えたLinuxシステムが必要です。さらに、Brakerは、次のCPAN-PERLモジュールをインストールする必要があります。
File::Spec::Functions
Hash::Merge
List::Util
MCE::Mutex
Module::Load::Conditional
Parallel::ForkManager
POSIX
Scalar::Util::Numeric
YAML
Math::Utils
File::HomeDir
GeneMark-ETPの場合、タンパク質とRNA-seqが提供されたときに使用されます。
YAML::XSData::DumperThread::Queuethreadsたとえば、ubuntuでは、cpanminus f4 : sudo cpanm Module::Name 、例えばsudo cpanm Hash::Mergeでモジュールをインストールします。
Brakerは、CPANでは利用できないPerlモジュールhelpMod_braker.pmも使用しています。このモジュールはBreakerリリースの一部であり、個別のインストールを必要としません。
Linuxマシンにルートアクセス許可がない場合は、次のようにAnaconda (https://www.anaconda.com/distribution/)環境をセットアップしてみてください。
wget https://repo.anaconda.com/archive/Anaconda3-2018.12-Linux-x86_64.sh
bash bin/Anaconda3-2018.12-Linux-x86_64.sh # do not install VS (needs root privileges)
conda install -c anaconda perl
conda install -c anaconda biopython
conda install -c bioconda perl-app-cpanminus
conda install -c bioconda perl-file-spec
conda install -c bioconda perl-hash-merge
conda install -c bioconda perl-list-util
conda install -c bioconda perl-module-load-conditional
conda install -c bioconda perl-posix
conda install -c bioconda perl-file-homedir
conda install -c bioconda perl-parallel-forkmanager
conda install -c bioconda perl-scalar-util-numeric
conda install -c bioconda perl-yaml
conda install -c bioconda perl-class-data-inheritable
conda install -c bioconda perl-exception-class
conda install -c bioconda perl-test-pod
conda install -c bioconda perl-file-which # skip if you are not comparing to reference annotation
conda install -c bioconda perl-mce
conda install -c bioconda perl-threaded
conda install -c bioconda perl-list-util
conda install -c bioconda perl-math-utils
conda install -c bioconda cdbtools
conda install -c eumetsat perl-yaml-xs
conda install -c bioconda perl-data-dumper
その後、Conda環境にいる間にBreakerやその他のソフトウェアを「いつものように」インストールします。注: Bioconda BreakerパッケージとBioconda Augustusパッケージがあります。彼らは働きます。しかし、それらは通常、GitHub上の両方のツールの開発コードに遅れをとっています。したがって、最新のソースの手動インストールと使用をお勧めします。
Breakerは、PerlおよびPythonスクリプトとPerlモジュールのコレクションです。 Breakerを実行するために呼び出される主なスクリプトは、 braker.plです。追加のPerlおよびPythonコンポーネントは次のとおりです。
align2hints.pl
filterGenemark.pl
filterIntronsFindStrand.pl
startAlign.pl
helpMod_braker.pm
findGenesInIntrons.pl
downsample_traingenes.pl
ensure_n_training_genes.py
get_gc_content.py
get_etp_hints.py
Breakerの一部であるすべてのスクリプト( *.plおよび*.pyで終了するファイル)は、Breakerを実行するために実行可能でなければなりません。 GithubからBreakerをダウンロードする場合、これはすでに当てはまるはずです。 USBスティックのブレイカーを別のコンピューターに転送する場合、実行可能性が上書きされる場合があります。必要なファイルが実行可能かどうかを確認するには、Braker Perlスクリプトを含むディレクトリに次のコマンドを実行します。
ls -l *.pl *.py
出力はこれに似ている必要があります。
-rwxr-xr-x 1 katharina katharina 18191 Mai 7 10:25 align2hints.pl
-rwxr-xr-x 1 katharina katharina 6090 Feb 19 09:35 braker_cleanup.pl
-rwxr-xr-x 1 katharina katharina 408782 Aug 17 18:24 braker.pl
-rwxr-xr-x 1 katharina katharina 5024 Mai 7 10:25 downsample_traingenes.pl
-rwxr-xr-x 1 katharina katharina 5024 Mai 7 10:23 ensure_n_training_genes.py
-rwxr-xr-x 1 katharina katharina 4542 Apr 3 2019 filter_augustus_gff.pl
-rwxr-xr-x 1 katharina katharina 30453 Mai 7 10:25 filterGenemark.pl
-rwxr-xr-x 1 katharina katharina 5754 Mai 7 10:25 filterIntronsFindStrand.pl
-rwxr-xr-x 1 katharina katharina 7765 Mai 7 10:25 findGenesInIntrons.pl
-rwxr-xr-x 1 katharina katharina 1664 Feb 12 2019 gatech_pmp2hints.pl
-rwxr-xr-x 1 katharina katharina 2250 Jan 9 13:55 log_reg_prothints.pl
-rwxr-xr-x 1 katharina katharina 4679 Jan 9 13:55 merge_transcript_sets.pl
-rwxr-xr-x 1 katharina katharina 41674 Mai 7 10:25 startAlign.pl
-rwxr-xr-xのxが各スクリプトに存在することが重要です。そうでない場合は、実行してください
`chmod a+x *.pl *.py`
ファイル属性を変更するため。
Braker Perlスクリプトが$PATH環境変数に存在するディレクトリを追加することが役立つ場合があります。単一のbashセッションについては、次のことを入力します。
PATH=/your_path_to_braker/:$PATH
export PATH
この$PATH変更をすべてのBASHセッションで利用できるようにするには、上記の行を起動スクリプト(例~/.bashrc )に追加します。
Breakerは、Breakerの一部ではないさまざまなバイオインフォマティクスソフトウェアツールを呼び出します。一部のツールは必須です。つまり、これらのツールがシステムに存在しない場合、ブレイカーはまったく実行されません。他のツールはオプションです。選択したモードでブレーカーを実行するために必要なすべてのツールをインストールしてください。
http://github.com/gatech-genemark/genemark-etpまたはhttps://topaz.gatech.edu/genemark/etp.for_braker.tar.gzからgenemark-etp f1をダウンロードしてください。 GeneMark-ETPのREADMEファイルで説明されているように、GeneMark-ETPを開梱してインストールします。
$PATH変数に既に含まれている場合、Brakerはgmes_petap.plまたはgmetp.plの場所を自動的に推測します。それ以外の場合、Brakerは、環境変数GENEMARK_PATHにそれらを検索するか、コマンドライン引数( --GENEMARK_PATH=/your_path_to_GeneMark_executables/ )でそれらを検索することにより、Genemark-es/et/ep/etp実行可能ファイルを見つけることができます。
現在のBASHセッションの環境変数を設定するには、以下を入力してください。
export GENEMARK_PATH=/your_path_to_GeneMark_executables/
上記の行をスタートアップスクリプト( ~/.bashrc )に追加して、すべてのBashセッションで利用できるようにします。
GeneMark-ES/ET/EP/ETP内のPERLスクリプトは、 /usr/bin/perlのデフォルトのperl位置で構成されています。
Anaconda環境でGeneMark-ES/ET/EP/ETPを実行している場合(または、他の理由で$PATH変数からPERLを使用する場合)、すべてのGeneMark-ES/ET/EP/ETPスクリプトのシバンを変更しますGenemark-ES/ET/EP/ETPフォルダー内にある次のコマンド:
perl change_path_in_perl_scripts.pl "/usr/bin/env perl"
check_install.bashを実行したり、 GeneMark-E-testsディレクトリで例を実行したりすることにより、GeneMark-ES/ET/EPが適切にインストールされているかどうかを確認できます。
GeneMark-ETPは下向きに互換性があります。つまり、BreakerのGeneMark-EPとGeneMark-ETの機能もカバーしています。
https://github.com/gaius-augustus/augustusのマスターブランチからAugustusをダウンロードしてください。 Augustusを開梱し、Augustus README.TXTに従ってアウグストゥスをインストールします。他のソースからの時代遅れのアウグストゥスバージョンを使用しないでください。たとえば、DebianパッケージやBiocondaパッケージなどです! Breakerは特に最新のAugustus/Scriptsディレクトリに大きく依存しており、他のソースが遅れていることがよくあります。
アウグストゥスが使用するライブラリのバージョンの問題を回避するために、自分のシステムにアウグストゥスをコンパイする必要があります。コンパイルの指示は、Augustus README.TXTファイル( Augustus/README.txt )に記載されています。
アウグストゥスは、 augustus 、遺伝子予測ツール、 Augustus/auxprogsにある追加のC ++ツール、およびAugustus/scriptsにあるPerlスクリプトで構成されています。 PERLスクリプトは実行可能である必要があります(セクションBreakerコンポーネントの指示を参照してください。
C ++ツールbam2hints RNA-seqで実行する場合のBreakerの重要なコンポーネントです。ソースはAugustus/auxprogs/bam2hintsにあります。システムにbam2hintsコンパイルすることを確認してください(Augustusがコンパイルされたときに自動的にコンパイルする必要がありますが、 bam2hintsの問題が発生した場合は、 Augustus/auxprogs/bam2hints/READMEの手順のトラブルシューティングをお読みください)。
BrakerはAugustusを訓練するパイプラインであるため、IEは種固有のパラメーターファイルを書き込みます。Breakerは、そのようなファイルを含むAugustusの構成ディレクトリへのアクセスを書き込む必要があります( Augustus/config/ )。システムにグローバルにAugustusをインストールする場合、通常、すべてのユーザーがconfigフォルダーを手紙を作成しません。 config Augustusのユーザーに再帰的に執筆可能なディレクトリを作成するか、ユーザーが許可を書いている場所にconfig/フォルダーを(再帰的に)コピーします。
Augustusは、環境変数$AUGUSTUS_CONFIG_PATHを探すことにより、 configフォルダーを見つけます。 $AUGUSTUS_CONFIG_PATH環境変数が設定されていない場合、BrakerはAugustusの実行可能ファイルを見つけたディレクトリに対してパス../configを調べます。または、Braker( --AUGUSTUS_CONFIG_PATH=/your_path_to_AUGUSTUS/Augustus/config/ )へのコマンドライン引数として変数を提供できます。現在のバッシュセッションの変数EGをエクスポートすることをお勧めします。
export AUGUSTUS_CONFIG_PATH=/your_path_to_AUGUSTUS/Augustus/config/
すべてのBashセッションで変数を使用できるようにするには、上記の行をスタートアップスクリプト( ~/.bashrcに追加します。
AugustusをDebianパッケージとしてインストールする場合は、Dockerfileをご覧ください。したがって、多くのスクリプトを修正する必要があります。
Brakerは、 $AUGUSTUS_CONFIG_PATHのAugustusのconfigディレクトリ全体、つまりその内容(少なくともgeneric )とextrinsicのサブフォルダーspecies期待しています! $AUGUSTUS_CONFIG_PATHで書く可能性のあるが空のフォルダーを提供することは、Breakerでは機能しません。 Augustus Binaryと$AUGUSTUS_CONFIG_PATHを分離する必要がある場合は、執筆不可能な構成コンテンツを書き込み可能な場所に再帰的にコピーすることをお勧めします。
Augustus at /usr/bin/augustus Augustusのシステム全体のインストールがある場合、 config sits at /usr/bin/augustus_config/ 。フォルダ/home/yours/あなたに手紙を書きます。次のコマンドでコピーします(さらに、当時の必要な変数を設定します)。
cp -r /usr/bin/Augustus/config/ /home/yours/
export AUGUSTUS_CONFIG_PATH=/home/yours/augustus_config
export AUGUSTUS_BIN_PATH=/usr/bin
export AUGUSTUS_SCRIPTS_PATH=/usr/bin/augustus_scripts
Augustusのバイナリとスクリプトのディレクトリを$PATH変数に追加すると、システムがこれらのツールを自動的に見つけることができます。 Brakerは別の環境変数( $AUGUSTUS_CONFIG_PATH )の場所からそれらを推測しようとするか、両方のディレクトリをbraker.plにコマンドライン引数として提供することを試みるため、Brakerを実行するための要件ではありませんが、それらを$PATH変数に追加します。現在のBASHセッションの場合、タイプ:
PATH=:/your_path_to_augustus/bin/:/your_path_to_augustus/scripts/:$PATH
export PATH
すべてのバッシュセッションについて、上記の行をスタートアップスクリプトに追加します(例~/.bashrc )。
Ubuntuでは、通常、Python3はデフォルトでインストールされ、 python3デフォルトで$PATH変数になり、Brakerは自動的に見つけます。ただし、 python3バイナリの位置を他の2つの方法で指定するオプションがあります。
環境変数をエクスポートする$PYTHON3_PATH 、例えば~/.bashrcファイル:
export PYTHON3_PATH=/path/to/python3/
コマンドラインオプションを指定します--PYTHON3_PATH=/path/to/python3/ to braker.pl 。
bamtoolsをダウンロードします(例: git clone https://github.com/pezmaster31/bamtools.git )。シェルに以下を入力してBamtoolsをインストールしてください。
cd your-bamtools-directory mkdir build cd build cmake .. make
すでに$PATH変数にある場合、Brakerは自動的にBamtoolsを見つけます。それ以外の場合、Brakerは、環境変数$BAMTOOLS_PATH使用するか、コマンドライン引数( --BAMTOOLS_PATH=/your_path_to_bamtools/bin/ f6 )を使用することにより、bamtoolsバイナリを見つけることができます。現在のBASHセッションの環境変数EGを設定するには、次のように入力してください。
export BAMTOOLS_PATH=/your_path_to_bamtools/bin/
上記の行をスタートアップスクリプト(例: ~/.bashrc )に追加して、すべてのバッシュセッションの環境変数を設定します。
NCBI Blast+またはダイヤモンドのいずれかを使用して、冗長トレーニング遺伝子を除去できます。両方のツールは必要ありません。ダイヤモンドが存在する場合、はるかに高速であるため、好まれます。
次のようにダイヤモンドを取得して開梱します。
wget http://github.com/bbuchfink/diamond/releases/download/v0.9.24/diamond-linux64.tar.gz
tar xzf diamond-linux64.tar.gz
すでに$PATH変数にある場合、Brakerは自動的にダイヤモンドを見つけます。それ以外の場合、Breakerは、環境変数$DIAMOND_PATH使用するか、コマンドライン引数( --DIAMOND_PATH=/your_path_to_diamond )を使用することにより、ダイヤモンドバイナリを見つけることができます。現在のBASHセッションの環境変数EGを設定するには、次のように入力してください。
export DIAMOND_PATH=/your_path_to_diamond/
上記の行をスタートアップスクリプト(例: ~/.bashrc )に追加して、すべてのバッシュセッションの環境変数を設定します。
Blast+を決定する場合は、 sudo apt-get install ncbi-blast+ blast+をインストールします。
すでに$PATH変数にある場合、Brakerは自動的にBLASTPを見つけます。それ以外の場合、Brakerは、環境変数$BLAST_PATH使用するか、コマンドライン引数( --BLAST_PATH=/your_path_to_blast/ )を使用することにより、blastpバイナリを見つけることができます。現在のBASHセッションの環境変数EGを設定するには、次のように入力してください。
export BLAST_PATH=/your_path_to_blast/
上記の行をスタートアップスクリプト(例: ~/.bashrc )に追加して、すべてのバッシュセッションの環境変数を設定します。
次のツールはGeneMark-ETPで必要であり、 $PATH変数にそれらを見つけようとします。したがって、 $PATHに場所を追加してください。
export PATH=$PATH:/your/path/to/Tool
以下のすべてのツールについて、上記の行をスタートアップスクリプト(例~/.bashrc )に追加して、すべてのbashセッションの$PATH変数を拡張します。
これらのソフトウェアツールは、RNA-seqとタンパク質データを使用してBreakerを実行する場合にのみ必須です!
stringtie2は、genemark-etpによって整列したRNA-seqアライメントを組み立てるために使用されます。 stringtie2の事前コンパイルバージョンは、https://ccb.jhu.edu/software/stringtie/#installからダウンロードできます。
RNA-seqとタンパク質データの両方でBreakerを実行する場合、Software Package BedToolsはGeneMark-ETPで必要です。 https://github.com/arq5x/bedtools2/releasesからbedtoolsをダウンロードできます。ここでは、事前コンパイルされたバージョンbedtools.static.binaryをダウンロードできます。
wget https://github.com/arq5x/bedtools2/releases/download/v2.30.0/bedtools.static.binary
mv bedtools.static.binary bedtools
chmod a+x
または、 bedtools-2.30.0.tar.gzをダウンロードして、 make 、Egを使用してソースからコンパイルすることもできます。
wget https://github.com/arq5x/bedtools2/releases/download/v2.30.0/bedtools-2.30.0.tar.gz
tar -zxvf bedtools-2.30.0.tar.gz
cd bedtools2
make
詳細については、https://bedtools.readthedocs.io/en/latest/content/installation.htmlを参照してください。
GFFREADは、GeneMark-ETPが必要とするユーティリティソフトウェアです。 https://github.com/gpertea/gffread/releases/download/v0.12.7/gffread-0.12.7.linux_x86_64.tar.gzからダウンロードでき、 make 、たとえばMakeでインストールできます。
wget https://github.com/gpertea/gffread/releases/download/v0.12.7/gffread-0.12.7.Linux_x86_64.tar.gz
tar xzf gffread-0.12.7.Linux_x86_64.tar.gz
cd gffread-0.12.7.Linux_x86_64
make
Samtoolsは、すべてのファイルが正しくフォーマットされている場合、GeneMark-ETPなしでBreakerを実行するためには必要ありません(つまり、すべてのシーケンスには短くユニークなFASTA名が必要です)。すべてのファイルが正しくfマットされているかどうかわからない場合は、Samtoolsを使用して特定の形式の問題を自動的に修正できるため、Samtoolsをインストールすると役立つ場合があります。
samtoolsの前提条件として、 htslibダウンロードしてインストールします(例: git clone https://github.com/samtools/htslib.git 、インストールについてはhtslibドキュメントに従ってください)。
samtools( git clone git://github.com/samtools/samtools.gitなど)をダウンロードしてインストールし、その後、インストールのためにSamtoolsドキュメントに従ってください)。
すでに$PATH変数にある場合、BrakerはSamtoolsを自動的に見つけます。それ以外の場合、Brakerは、コマンドラインの引数( --SAMTOOLS_PATH=/your_path_to_samtools/ )を取得するか、環境変数$SAMTOOLS_PATH使用することにより、Samtoolsを見つけることができます。変数をエクスポートするには、例えば現在のbashセッションでは、次のように入力してください。
export SAMTOOLS_PATH=/your_path_to_samtools/
上記の行をスタートアップスクリプト(例: ~/.bashrc )に追加して、すべてのバッシュセッションの環境変数を設定します。
Biopythonがインストールされている場合、Breakerは、Augustusによって予測されたコーディングシーケンスとタンパク質配列を備えたFasta-Filesを生成し、MakeHub R16を使用してBreaker Runの視覚化のためにトラックデータハブを生成できます。これらはオプションの手順です。 1つ目は、コマンドラインフラグ--skipGetAnnoFromFastaで無効にすることができます。2番目は、コマンドラインオプション--makehub [email protected]使用してアクティブ化できます。実行されます。
ubuntuで、次のようにpython3パッケージマネージャーをインストールします
`sudo apt-get install python3-pip`
次に、次のようにBioPythonをインストールします。
`sudo pip3 install biopython`
CdbfastaとCdbyankは、Augustus Script fix_in_frame_stop_codon_genes.pyを使用して、フレームストップコドン(スプライス停止コドン)でアウグストゥスの遺伝子を補正するためにBrakerが必要としています。これは、 --skip_fixing_broken_genesでスキップできます。
ubuntuで、次のようにcdbfastaをインストールします
sudo apt-get install cdbfasta
他のシステムの場合、たとえばhttps://github.com/gpertea/cdbfastaからcdbfastaを取得できます。
git clone https://github.com/gpertea/cdbfasta.git
cd cdbfasta
make all
Ubuntuでは、CDBFASTAとCDBYANKがインストール後に$PATH変数になり、Breakerは自動的に見つけます。ただし、他の2つの方法でcdbfastaとcdbyankバイナリの場所を指定するオプションがあります。
$CDBTOOLS_PATH 、例えば~/.bashrcファイル: export CDBTOOLS_PATH=/path/to/cdbtools/
--CDBTOOLS_PATH=/path/to/cdbtools/ to braker.plを指定します。 注: Braker内のスタンドアロンSpaln(ProthintのOouside)のサポートは非推奨です。
このツールは、Prothintを実行する場合、またはProthintの外側でSpalnを使用してBreakerとゲノムアライメントにタンパク質を実行したい場合に必要です。 Prothintの外側でSpalnを使用することは、ターゲットゲノムまでの短い進化距離の注釈付き種が利用可能な場合にのみ、適切なアプローチです。 BrakerのProthintを介してSpalnを実行することをお勧めします。 Prothintはスパルンバイナリをもたらします。それがシステムで動作しない場合は、https://github.com/ogotoh/spalnからSpalnをダウンロードしてください。 spaln/doc/SpalnReadMe22.pdfに従って開梱してインストールします。
Brakerは、環境変数$ALIGNMENT_TOOL_PATH使用して、Spaln実行可能ファイルを見つけようとします。あるいは、これはコマンドライン引数( --ALIGNMENT_TOOL_PATH=/your/path/to/spaln )として提供することができます。
このツールは、予測遺伝子にUTR(RNA-seqデータから)を追加したい場合、またはAugustusのUTRパラメーターをトレーニングし、UTRで遺伝子を予測する場合にのみ必要です。いずれにせよ、GushrはRNA-seqデータの入力を必要とします。
Gushrはhttps://github.com/gaius-augustus/gushrでダウンロードできます。入力して取得します。
git clone https://github.com/Gaius-Augustus/GUSHR.git
GushrはGemoma JarファイルR19、 R20、 R21を実行し、このJARファイルにはJava 1.8が必要です。 Ubuntuでは、次のコマンドでJava 1.8をインストールできます。
sudo apt-get install openjdk-8-jdk
システムにいくつかのJavaバージョンがインストールされている場合は、実行してJavaでBrakerを使用して1.8を実行できることを確認してください
sudo update-alternatives --config java
正しいバージョンを選択します。
Switch --UTR=on 、bamtowig.pyには、http://hgdownload.soe.ucsc.edu/admin/exeからダウンロードできる次のツールが必要になります。
TwoBitinfo
fatotwobit
これらのツールを$パスにインストールすることはオプションです。そうでない場合、そして--UTR=on切り替えた場合、bamtowig.pyはそれらをワーキングディレクトリに自動的にダウンロードします。
Automaticalyを作成したい場合は、Braker Runのトラックデータハブを生成したい場合は、https://github.com/gaius-augustus/makehubで入手できるMakeHubソフトウェアが必要です。ソフトウェアをダウンロードします( git clone https://github.com/Gaius-Augustus/MakeHub.gitを実行するか、https://github.com/githus-augustus/makehub/releasesからリリースを選択します。パッケージリリースをダウンロードした場合( unzip MakeHub.zipまたはtar -zxvf MakeHub.tar.gzなど。
Brakerは、環境変数$MAKEHUB_PATHを使用して、make_hub.pyスクリプトを見つけようとします。あるいは、これはコマンドライン引数( --MAKEHUB_PATH=/your/path/to/MakeHub/ )として提供できます。 Breakerは、システム上のMakeHubの場所を推測しようとすることもできます。
NCBIのSRAからRNA-SEQライブラリをダウンロードしたい場合は、SRAツールキットが必要です。 http://daehwankimlab.github.io/hisat2/download/#version-hisat2-221からSRA Toolkitの事前コンパイルバージョンを入手できます。
Breakerは、環境変数$SRATOOLS_PATH使用して、SRA Toolkit(FastQ-Dump、Prefetch)から実行可能なバイナリを見つけようとします。あるいは、これはコマンドライン引数( --SRATOOLS_PATH=/your/path/to/SRAToolkit/ )として提供することができます。 Breakerは、実行可能ファイルが$PATH変数にある場合、システム上のSRAツールキットの場所を推測しようとすることもできます。
整列していないRNA-SEQ読み取りを使用する場合は、HISAT2ソフトウェアをゲノムにマッピングする必要があります。 HISAT2の事前補償バージョンは、http://daehwankimlab.github.io/hisat2/download/#version-hisat2-221からダウンロードできます。
Breakerは、環境変数$HISAT2_PATHを使用して、実行可能なHISAT2バイナリ(HISAT2、HISAT2-BUILD)を見つけようとします。あるいは、これはコマンドライン引数( --HISAT2_PATH=/your/path/to/HISAT2/ )として提供できます。 Breakerは、実行可能ファイルが$PATH変数にある場合、システム上のHISAT2の位置を推測しようとすることもできます。
Buscoの完全性の最大化モードでBraker内でTsebraを実行したい場合は、CompleSMをインストールする必要があります。
wget https://github.com/huangnengCSU/compleasm/releases/download/v0.2.4/compleasm-0.2.4_x64-linux.tar.bz2
tar -xvjf compleasm-0.2.4_x64-linux.tar.bz2 &&
結果のフォルダーを$PATH変数にcompleasm_kitを追加します。
export PATH=$PATH:/your/path/to/compleasm_kit
CompleaSMにはパンダが必要です。
pip install pandas
Braker(Braker.pl)は、GetConfを使用して、システムで実行できるスレッドの数を確認します。 ubuntuでは、次のようにインストールできます。
sudo apt-get install libc-bin
以下では、「典型的な」ブレーカーがさまざまな入力データ型を求める呼びかけについて説明します。一般に、繰り返しのためにソフトマスクされたゲノムシーケンスでブレーカーを実行することをお勧めします。ブレーカーは、繰り返しのためにソフトマスクされたゲノムにのみ適用する必要があります!
This approach is suitable for genomes of species for which RNA-Seq libraries with good transcriptome coverage are available and for which protein data is not at hand. The pipeline is illustrated in Figure 2.
BRAKER has several ways to receive RNA-Seq data as input:
You can provide ID(s) of RNA-Seq libraries from SRA (in case of multiple IDs, separate them by comma) as argument to --rnaseq_sets_ids . The libraries belonging to the IDs are then downloaded automatically by BRAKER, eg:
braker.pl --species=yourSpecies --genome=genome.fasta
--rnaseq_sets_ids=SRA_ID1,SRA_ID2
You can use local FASTQ file(s) of unaligned reads as input. In this case, you have to provide BRAKER with the ID(s) of the RNA-Seq set(s) as argument to --rnaseq_sets_ids and the path(s) to the directories, where the FASTQ files are located as argument to --rnaseq_sets_dirs . For each ID ID , BRAKER will search in these directories for one FASTQ file named ID.fastq if the reads are unpaired, or for two FASTQ files named ID_1.fastq and ID_2.fastq if they are paired.
For example, if you have a paired library called 'SRA_ID1' and an unpaired library named 'SRA_ID2', you have to have a directory /path/to/local/fastq/files/ , where the files SRA_ID1_1.fastq , SRA_ID1_2.fastq , and SRA_ID2.fastq reside. Then, you could run BRAKER with following command:
braker.pl --species=yourSpecies --genome=genome.fasta
--rnaseq_sets_ids=SRA_ID1,SRA_ID2
--rnaseq_sets_dirs=/path/to/local/fastq/files/
There are two ways of supplying BRAKER with RNA-Seq data as bam file(s). First, you can do it in the same way as you would supply FASTQ file(s): Provide the ID(s)/name(s) of your bam file(s) as argument to --rnaseq_sets_ids and specify directories where the bam files reside with --rnaseq_sets_dirs . BRAKER will automatically detect that these ID(s) are bam and not FASTQ file(s), eg:
braker.pl --species=yourSpecies --genome=genome.fasta
--rnaseq_sets_ids=BAM_ID1,BAM_ID2
--rnaseq_sets_dirs=/path/to/local/bam/files/
Second, you can specify the paths to your bam file(s) directly, eg can either extract RNA-Seq spliced alignment information from bam files, or it can use such extracted information, directly.
braker.pl --species=yourSpecies --genome=genome.fasta
--bam=file1.bam,file2.bam
Please note that we generally assume that bam files were generated with HiSat2 because that is the aligner that would also be executed by BRAKER3 with fastq input. If you want for some reason to generate the bam files with STAR, use the option --outSAMstrandField intronMotif of STAR to produce files that are compatible wiht StringTie in BRAKER3.
In order to run BRAKER with RNA-Seq spliced alignment information that has already been extracted, run:
braker.pl --species=yourSpecies --genome=genome.fasta
--hints=hints1.gff,hints2.gff
The format of such a hints file must be as follows (tabulator separated file):
chrName b2h intron 6591 8003 1 + . pri=4;src=E
chrName b2h intron 6136 9084 11 + . mult=11;pri=4;src=E
...
The source b2h in the second column and the source tag src=E in the last column are essential for BRAKER to determine whether a hint has been generated from RNA-Seq data.
It is also possible to provide RNA-Seq sets in different ways for the same BRAKER run, any combination of above options is possible. It is not recommended to provide RNA-Seq data with --hints if you run BRAKER in ETPmode (RNA-Seq and protein data), because GeneMark-ETP won't use these hints!
This approach is suitable for genomes of species for which no RNA-Seq libraries are available. A large database of proteins (with possibly longer evolutionary distance to the target species) should be used in this case. This mode is illustrated in figure 9.
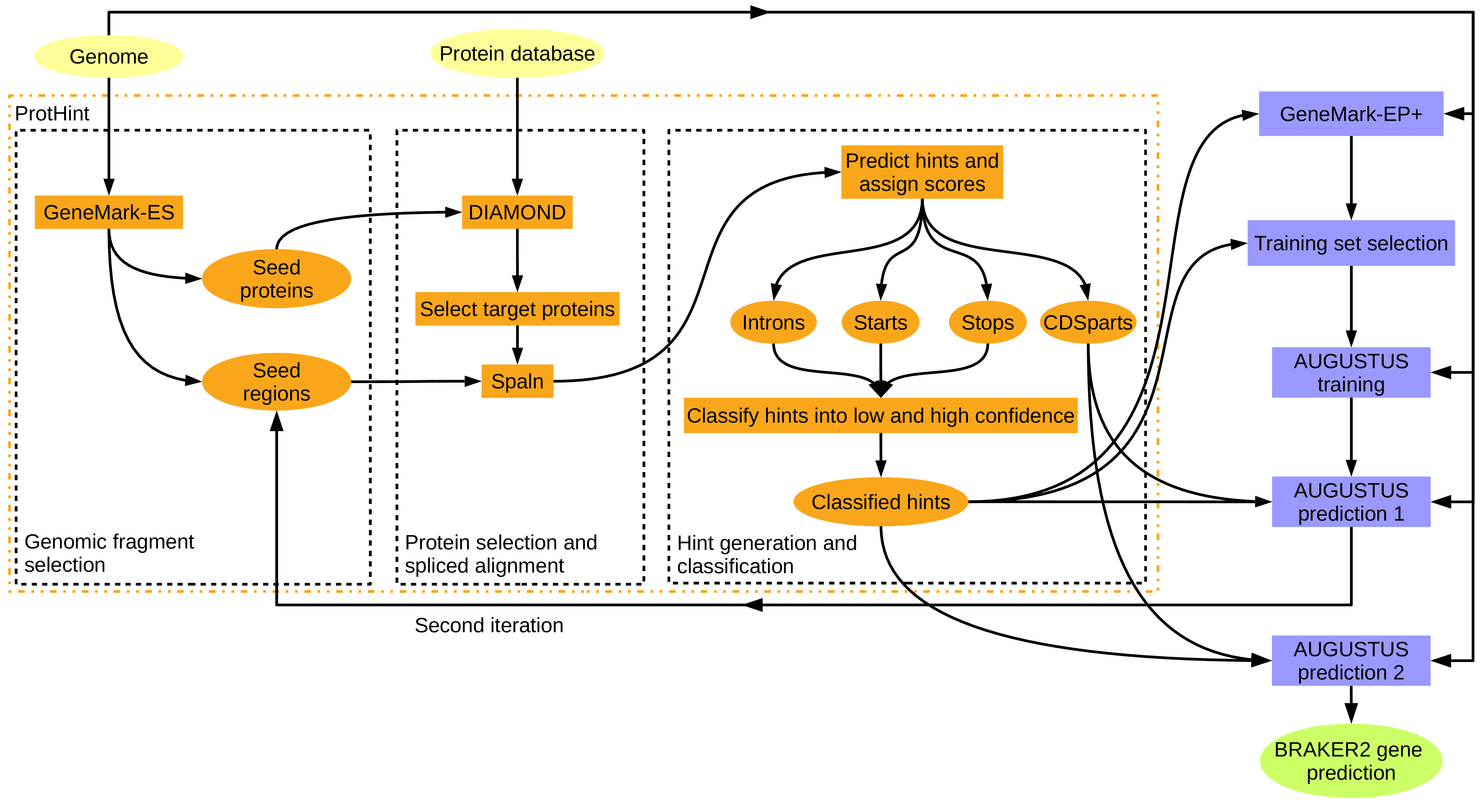
Figure 9: BRAKER with proteins of any evolutionary distance. ProtHint protein mapping pipelines is used to generate protein hints. ProtHint automatically determines which alignments are from close relatives, and which are from rather distant relatives.
For running BRAKER in this mode, type:
braker.pl --genome=genome.fa --prot_seq=proteins.fa
We recommend using OrthoDB as basis for proteins.fa . The instructions on how to prepare the input OrthoDB proteins are documented here: https://github.com/gatech-genemark/ProtHint#protein-database-preparation.
You can of course add additional protein sequences to that file, or try with a completely different database. Any database will need several representatives for each protein, though.
Instead of having BRAKER run ProtHint, you can also start BRAKER with hints already produced by ProtHint, by providing ProtHint's prothint_augustus.gff output:
braker.pl --genome=genome.fa --hints=prothint_augustus.gff
The format of prothint_augustus.gff in this mode looks like this:
2R ProtHint intron 11506230 11506648 4 + . src=M;mult=4;pri=4
2R ProtHint intron 9563406 9563473 1 + . grp=69004_0:001de1_702_g;src=C;pri=4;
2R ProtHint intron 8446312 8446371 1 + . grp=43151_0:001cae_473_g;src=C;pri=4;
2R ProtHint intron 8011796 8011865 2 - . src=P;mult=1;pri=4;al_score=0.12;
2R ProtHint start 234524 234526 1 + . src=P;mult=1;pri=4;al_score=0.08;
The prediction of all hints with src=M will be enforced. Hints with src=C are 'chained evidence', ie they will only be incorporated if all members of the group (grp=...) can be incorporated in a single transcript. All other hints have src=P in the last column. Supported features in column 3 are intron , start , stop and CDSpart .
If RNA-Seq (and only RNA-Seq) data is provided to BRAKER as a bam-file, and if the genome is softmasked for repeats, BRAKER can automatically train UTR parameters for AUGUSTUS. After successful training of UTR parameters, BRAKER will automatically predict genes including coverage information form RNA-Seq data. Example call:
braker.pl --species=yourSpecies --genome=genome.fasta
--bam=file.bam --UTR=on
Warnings:
This feature is experimental!
--UTR=on is currently not compatible with bamToWig.py as released in AUGUSTUS 3.3.3; it requires the current development code version from the github repository (git clone https://github.com/Gaius-Augustus/Augustus.git).
--UTR=on increases memory consumption of AUGUSTUS. Carefully monitor jobs if your machine was close to maxing RAM without --UTR=on! Reducing the number of cores will also reduce RAM consumption.
UTR prediction sometimes improves coding sequence prediction accuracy, but not always. If you try this feature, carefully compare results with and without UTR parameters, afterwards (eg in UCSC Genome Browser).
For running BRAKER without UTR parameters, it is not very important whether RNA-Seq data was generated by a stranded protocol (because spliced alignments are 'artificially stranded' by checking the splice site pattern). However, for UTR training and prediction, stranded libraries may provide information that is valuable for BRAKER.
After alignment of the stranded RNA-Seq libraries, separate the resulting bam file entries into two files: one for plus strand mappings, one for minus strand mappings. Call BRAKER as follows:
braker.pl --species=yourSpecies --genome=genome.fasta
--bam=plus.bam,minus.bam --stranded=+,-
--UTR=on
You may additionally include bam files from unstranded libraries. Those files will not used for generating UTR training examples, but they will be included in the final gene prediction step as unstranded coverage information, example call:
braker.pl --species=yourSpecies --genome=genome.fasta
--bam=plus.bam,minus.bam,unstranded.bam
--stranded=+,-,. --UTR=on
Warning: This feature is experimental and currently has low priority on our maintenance list!
The native mode for running BRAKER with RNA-Seq and protein data. This will call GeneMark-ETP, which will use RNA-Seq and protein hints for training GeneMark-ETP. Subsequently, AUGUSTUS is trained on 'high-confindent' genes (genes with very high extrinsic evidence support) from the GeneMark-ETP prediction and a set of genes is predicted by AUGUSTUS. In a last step, the predictions of AUGUSTUS and GeneMark-ETP are combined using TSEBRA.
Alignment of RNA-Seq reads
GeneMark-ETP utilizes Stringtie2 to assemble RNA-Seq data, which requires that the aligned reads (BAM files) contain the XS (strand) tag for spliced reads. Therefore, if you align your reads with HISAT2, you must enable the --dta option, or if you use STAR, you must use the --outSAMstrandField intronMotif option. TopHat alignments include this tag by default.
To call the pipeline in this mode, you have to provide it with a protein database using --prot_seq (as described in BRAKER with protein data), and RNA-Seq data either by their SRA ID so that they are downloaded by BRAKER, as unaligned reads in FASTQ format, and/or as aligned reads in bam format (as described in BRAKER with RNA-Seq data). You could also specify already processed extrinsic evidence using the --hints option. However, this is not recommend for a normal BRAKER run in ETPmode, as these hints won't be used in the GeneMark-ETP step. Only use --hints when you want to skip the GenMark-ETP step!
Examples of how you could run BRAKER in ETPmode:
braker.pl --genome=genome.fa --prot_seq=orthodb.fa
--rnaseq_sets_ids=SRA_ID1,SRA_ID2
--rnaseq_sets_dirs=/path/to/local/RNA-Seq/files/
braker.pl --genome=genome.fa --prot_seq=orthodb.fa
--rnaseq_sets_ids=SRA_ID1,SRA_ID2,SRA_ID3
braker.pl --genome=genome.fa --prot_seq=orthodb.fa
--bam=/path/to/SRA_ID1.bam,/path/to/SRA_ID2.bam
A preliminary protocol for integration of assembled subreads from PacBio ccs sequencing in combination with short read Illumina RNA-Seq and protein database is described at https://github.com/Gaius-Augustus/BRAKER/blob/master/docs/long_reads/long_read_protocol .md
We forked GeneMark-ETP and hard coded that StringTie will perform long read assembly in that particular version. If you want to use this 'fast-hack' version for BRAKER, you have to prepare the BAM file with long read to genome spliced alignments outside of BRAKER, eg:
T=48 # adapt to your number of threads
minimap2 -t${T} -ax splice:hq -uf genome.fa isoseq.fa > isoseq.sam
samtools view -bS --threads ${T} isoseq.sam -o isoseq.bam
Pull the adapted container:
singularity build braker3_lr.sif docker://teambraker/braker3:isoseq
Calling BRAKER3 with a BAM file of spliced-aligned IsoSeq Reads:
singularity exec -B ${PWD}:${PWD} braker3_lr.sif braker.pl --genome=genome.fa --prot_seq=protein_db.fa –-bam=isoseq.bam --threads=${T}
Warning Do NOT mix short read and long read data in this BRAKER/GeneMark-ETP variant!
Warning The accuracy of gene prediction here heavily depends on the depth of your isoseq data. We verified with PacBio HiFi reads from 2022 that given sufficient completeness of the assembled transcriptome you will reach similar results as with short reads. However, we also observed a drop in accuracy compared to short reads when using other long read data sets with higher error rates and less sequencing depth.
Please run braker.pl --help to obtain a full list of options.
Compute AUGUSTUS ab initio predictions in addition to AUGUSTUS predictions with hints (additional output files: augustus.ab_initio.* . This may be useful for estimating the quality of training gene parameters when inspecting predictions in a Browser.
One or several command line arguments to be passed to AUGUSTUS, if several arguments are given, separate them by whitespace, ie "--first_arg=sth --second_arg=sth" . This may be be useful if you know that gene prediction in your particular species benefits from a particular AUGUSTUS argument during the prediction step.
Specifies the maximum number of threads that can be used during computation. BRAKER has to run some steps on a single thread, others can take advantage of multiple threads. If you use more than 8 threads, this will not speed up all parallelized steps, in particular, the time consuming optimize_augustus.pl will not use more than 8 threads. However, if you don't mind some threads being idle, using more than 8 threads will speed up other steps.
GeneMark-ETP option: run algorithm with branch point model. Use this option if you genome is a fungus.
Use the present config and parameter files if they exist for 'species'; will overwrite original parameters if BRAKER performs an AUGUSTUS training.
Execute CRF training for AUGUSTUS; resulting parameters are only kept for final predictions if they show higher accuracy than HMM parameters. This increases runtime!
Change the parameter
Generate UTR training examples for AUGUSTUS from RNA-Seq coverage information, train AUGUSTUS UTR parameters and predict genes with AUGUSTUS and UTRs, including coverage information for RNA-Seq as evidence. This is an experimental feature!
If you performed a BRAKER run without --UTR=on, you can add UTR parameter training and gene prediction with UTR parameters (and only RNA-Seq hints) with the following command:
braker.pl --genome=../genome.fa --addUTR=on
--bam=../RNAseq.bam --workingdir=$wd
--AUGUSTUS_hints_preds=augustus.hints.gtf
--threads=8 --skipAllTraining --species=somespecies
Modify augustus.hints.gtf to point to the AUGUSTUS predictions with hints from previous BRAKER run; modify flaning_DNA value to the flanking region from the log file of your previous BRAKER run; modify some_new_working_directory to the location where BRAKER should store results of the additional BRAKER run; modify somespecies to the species name used in your previous BRAKER run.
Add UTRs from RNA-Seq converage information to AUGUSTUS gene predictions using GUSHR. No training of UTR parameters and no gene prediction with UTR parameters is performed.
If you performed a BRAKER run without --addUTR=on, you can add UTRs results of a previous BRAKER run with the following command:
braker.pl --genome=../genome.fa --addUTR=on
--bam=../RNAseq.bam --workingdir=$wd
--AUGUSTUS_hints_preds=augustus.hints.gtf --threads=8
--skipAllTraining --species=somespecies
Modify augustus.hints.gtf to point to the AUGUSTUS predictions with hints from previous BRAKER run; modify some_new_working_directory to the location where BRAKER should store results of the additional BRAKER run; this run will not modify AUGUSTUS parameters. We recommend that you specify the original species of the original run with --species=somespecies . Otherwise, BRAKER will create an unneeded species parameters directory Sp_* .
If --UTR=on is enabled, strand-separated bam-files can be provided with --bam=plus.bam,minus.bam . In that case, --stranded=... should hold the strands of the bam files ( + for plus strand, - for minus strand, . for unstranded). Note that unstranded data will be used in the gene prediction step, only, if the parameter --stranded=... is set. This is an experimental feature! GUSHR currently does not take advantage of stranded data.
If --makehub and [email protected] (with your valid e-mail adress) are provided, a track data hub for visualizing results with the UCSC Genome Browser will be generated using MakeHub (https://github.com/Gaius-Augustus/MakeHub).
By default, GeneMark-ES/ET/EP/ETP uses a probability of 0.001 for predicting the donor splice site pattern GC (instead of GT). It may make sense to increase this value for species where this donor splice site is more common. For example, in the species Emiliania huxleyi , about 50% of donor splice sites have the pattern GC (https://media.nature.com/original/nature-assets/nature/journal/v499/n7457/extref/nature12221-s2.pdf, page 5).
Use a species-specific lineage, eg arthropoda_odb10 for an arthropod. BRAKER does not support auto-typing of the lineage.
Specifying a BUSCO-lineage invokes two changes in BRAKER R28 :
BRAKER will run compleasm with the specified lineage in genome mode and convert the detected BUSCO matches into hints for AUGUSTUS. This may increase the number of BUSCOs in the augustus.hints.gtf file slightly.
BRAKER will invoke best_by_compleasm.py to check whether the braker.gtf file that is by default generated by TSEBRA has the lowest amount of missing BUSCOs compared to the augustus.hints.gtf and the genemark.gtf file. If not, the following decision schema is applied to re-run TSEBRA to minimize the missing BUSCOs in the final output of BRAKER (always braker.gtf). If an alternative and better gene set is created, the original braker.gtf gene set is moved to a directory called braker_original. Information on what happened during the best_by_compleasm.py run is written to the file best_by_compleasm.log.
![best_by_busco[fig14]](https://images.downcodes.com/uploads/20250214/img_67aee79a11fd439.png)
Please note that using BUSCO to assess the quality of a gene set, in particular when comparing BRAKER to other pipelines, does not make sense once you specified a BUSCO lineage. We recommend that you use other measures to assess the quality of your gene set, eg by comparing it to a reference gene set or running OMArk.
BRAKER produces several important output files in the working directory.
braker.gtf: Final gene set of BRAKER. This file may contain different contents depending on how you called BRAKER
in ETPmode: Final gene set of BRAKER consisting of genes predicted by AUGUSTUS and GeneMark-ETP that were combined and filtered by TSEBRA.
otherwise: Union of augustus.hints.gtf and reliable GeneMark-ES/ET/EP predictions (genes fully supported by external evidence). In --esmode , this is the union of augustus.ab_initio.gtf and all GeneMark-ES genes. Thus, this set is generally more sensitive (more genes correctly predicted) and can be less specific (more false-positive predictions can be present). This output is not necessarily better than augustus.hints.gtf, and it is not recommended to use it if BRAKER was run in ESmode.
braker.codingseq: Final gene set with coding sequences in FASTA format
braker.aa: Final gene set with protein sequences in FASTA format
braker.gff3: Final gene set in gff3 format (only produced if the flag --gff3 was specified to BRAKER.
Augustus/*: Augustus gene set(s) in as gtf/conding/aa files
GeneMark-E*/genemark.gtf: Genes predicted by GeneMark-ES/ET/EP/EP+/ETP in GTF-format.
hintsfile.gff: The extrinsic evidence data extracted from RNAseq.bam and/or protein data.
braker_original/*: Genes predicted by BRAKER (TSEBRA merge) before compleasm was used to improve BUSCO completeness
bbc/*: output folder of best_by_compleasm.py script from TSEBRA that is used to improve BUSCO completeness in the final output of BRAKER
Output files may be present with the following name endings and formats:
Coding sequences in FASTA-format are produced if the flag --skipGetAnnoFromFasta was not set.
Protein sequence files in FASTA-format are produced if the flag --skipGetAnnoFromFasta was not set.
For details about gtf format, see http://www.sanger.ac.uk/Software/formats/GFF/. A GTF-format file contains one line per predicted exon.例:
HS04636 AUGUSTUS initial 966 1017 . + 0 transcript_id "g1.1"; gene_id "g1";
HS04636 AUGUSTUS internal 1818 1934 . + 2 transcript_id "g1.1"; gene_id "g1";
The columns (fields) contain:
seqname source feature start end score strand frame transcript ID and gene ID
If the --makehub option was used and MakeHub is available on your system, a hub directory beginning with the name hub_ will be created. Copy this directory to a publicly accessible web server. A file hub.txt resides in the directory. Provide the link to that file to the UCSC Genome Browser for visualizing results.
An incomplete example data set is contained in the directory BRAKER/example . In order to complete the data set, please download the RNA-Seq alignment file (134 MB) with wget :
cd BRAKER/example
wget http://topaz.gatech.edu/GeneMark/Braker/RNAseq.bam
In case you have trouble accessing that file, there's also a copy available from another server:
cd BRAKER/example
wget http://bioinf.uni-greifswald.de/augustus/datasets/RNAseq.bam
The example data set was not compiled in order to achieve optimal prediction accuracy, but in order to quickly test pipeline components. The small subset of the genome used in these test examples is not long enough for BRAKER training to work well.
Data corresponds to the last 1,000,000 nucleotides of Arabidopsis thaliana 's chromosome Chr5, split into 8 artificial contigs.
RNA-Seq alignments were obtained by VARUS.
The protein sequences are a subset of OrthoDB v10 plants proteins.
List of files:
genome.fa - genome file in fasta formatRNAseq.bam - RNA-Seq alignment file in bam format (this file is not a part of this repository, it must be downloaded separately from http://topaz.gatech.edu/GeneMark/Braker/RNAseq.bam)RNAseq.hints - RNA-Seq hints (can be used instead of RNAseq.bam as RNA-Seq input to BRAKER)proteins.fa - protein sequences in fasta formatThe below given commands assume that you configured all paths to tools by exporting bash variables or that you have the necessary tools in your $PATH.
The example data set also contains scripts tests/test*.sh that will execute below listed commands for testing BRAKER with the example data set. You find example results of AUGUSTUS and GeneMark-ES/ET/EP/ETP in the folder results/test* . Be aware that BRAKER contains several parts where random variables are used, ie results that you obtain when running the tests may not be exactly identical. To compare your test results with the reference ones, you can use the compare_intervals_exact.pl script as follows:
# Compare CDS features
compare_intervals_exact.pl --f1 augustus.hints.gtf --f2 ../../results/test${N}/augustus.hints.gtf --verbose
# Compare transcripts
compare_intervals_exact.pl --f1 augustus.hints.gtf --f2 ../../results/test${N}/augustus.hints.gtf --trans --verbose
Several tests use --gm_max_intergenic 10000 option to make the test runs faster. It is not recommended to use this option in real BRAKER runs, the speed increase achieved by adjusting this option is negligible on full-sized genomes.
We give runtime estimations derived from computing on Intel(R) Xeon(R) CPU E5530 @ 2.40GHz .
The following command will run the pipeline according to Figure 3:
braker.pl --genome genome.fa --bam RNAseq.bam --threads N --busco_lineage=lineage_odb10
This test is implemented in test1.sh , expected runtime is ~20 minutes.
The following command will run the pipeline according to Figure 4:
braker.pl --genome genome.fa --prot_seq proteins.fa --threads N --busco_lineage=lineage_odb10
This test is implemented in test2.sh , expected runtime is ~20 minutes.
The following command will run a pipeline that first trains GeneMark-ETP with protein and RNA-Seq hints and subsequently trains AUGUSTUS on the basis of GeneMark-ETP predictions. AUGUSTUS predictions are also performed with hints from both sources, see Figure 5.
Run with local RNA-Seq file:
braker.pl --genome genome.fa --prot_seq proteins.fa --bam ../RNAseq.bam --threads N --busco_lineage=lineage_odb10
This test is implemented in test3.sh , expected runtime is ~20 minutes.
Download RNA-Seq library from Sequence Read Archive (~1gb):
braker.pl --genome genome.fa --prot_seq proteins.fa --rnaseq_sets_ids ERR5767212 --threads N --busco_lineage=lineage_odb10
This test is implemented in test3_4.sh , expected runtime is ~35 minutes.
The training step of all pipelines can be skipped with the option --skipAllTraining . This means, only AUGUSTUS predictions will be performed, using pre-trained, already existing parameters. For example, you can predict genes with the command:
braker.pl --genome=genome.fa --bam RNAseq.bam --species=arabidopsis
--skipAllTraining --threads N
This test is implemented in test4.sh , expected runtime is ~1 minute.
The following command will run the pipeline with no extrinsic evidence:
braker.pl --genome=genome.fa --esmode --threads N
This test is implemented in test5.sh , expected runtime is ~20 minutes.
The following command will run BRAKER with training UTR parameters from RNA-Seq coverage data:
braker.pl --genome genome.fa --bam RNAseq.bam --UTR=on --threads N
This test is implemented in test6.sh , expected runtime is ~20 minutes.
The following command will add UTRs to augustus.hints.gtf from RNA-Seq coverage data:
braker.pl --genome genome.fa --bam RNAseq.bam --addUTR=on --threads N
This test is implemented in test7.sh , expected runtime is ~20 minutes.
There is currently no clean way to restart a failed BRAKER run (after solving some problem). However, it is possible to start a new BRAKER run based on results from a previous run -- given that the old run produced the required intermediate results. We will in the following refer to the old working directory with variable ${BRAKER_OLD} , and to the new BRAKER working directory with ${BRAKER_NEW} . The file what-to-cite.txt will always only refer to the software that was actually called by a particular run. You might have to combine the contents of ${BRAKER_NEW}/what-to-cite.txt with ${BRAKER_OLD}/what-to-cite.txt for preparing a publication. The following figure illustrates at which points BRAKER run may be intercepted.
![braker-intercept[fig8]](https://images.downcodes.com/uploads/20250214/img_67aee79a12cab310.png)
Figure 10: Points for intercepting a BRAKER run and reusing intermediate results in a new BRAKER run.
This option is only possible for BRAKER in ETmode or EPmode andないin ETPmode!
If you have access to an existing BRAKER output that contains hintsfiles that were generated from extrinsic data, such as RNA-Seq or protein sequences, you can recycle these hints files in a new BRAKER run. Also, hints from a separate ProtHint run can be directly used in BRAKER.
The hints can be given to BRAKER with --hints ${BRAKER_OLD}/hintsfile.gff option. This is illustrated in the test files test1_restart1.sh , test2_restart1.sh , test4_restart1.sh . The other modes (for which this test is missing) cannot be restarted in this way.
The GeneMark result can be given to BRAKER with --geneMarkGtf ${BRAKER_OLD}/GeneMark*/genemark.gtf option if BRAKER is run in ETmode or EPmode. This is illustrated in the test files test1_restart2.sh , test2_restart2.sh , test5_restart2.sh .
In ETPmode, you can either provide BRAKER with the results of the GeneMarkETP step manually, with --geneMarkGtf ${BRAKER_OLD}/GeneMark-ETP/proteins.fa/genemark.gtf , --traingenes ${BRAKER_OLD}/GeneMark-ETP/training.gtf , and --hints ${BRAKER_OLD}/hintsfile.gff (see test3_restart1.sh for an example), or you can specify the previous GeneMark-ETP results with the option --gmetp_results_dir ${BRAKER_OLD}/GeneMark-ETP/ so that BRAKER can search for the files automatically (see test3_restart2.sh for an example).
The trained species parameters for AGUSTUS can be passed with --skipAllTraining and --species $speciesName options. This is illustrated in test*_restart3.sh files. Note that in ETPmode you have to specify the GeneMark files as described in Option 2!
Before reporting bugs, please check that you are using the most recent versions of GeneMark-ES/ET/EP/ETP, AUGUSTUS and BRAKER. Also, check the list of Common problems, and the Issue list on GitHub before reporting bugs. We do monitor open issues on GitHub. Sometimes, we are unable to help you, immediately, but we try hard to solve your problems.
If you found a bug, please open an issue at https://github.com/Gaius-Augustus/BRAKER/issues (or contact [email protected] or [email protected]).
Information worth mentioning in your bug report:
Check in braker/yourSpecies/braker.log at which step braker.pl crashed.
There are a number of other files that might be of interest, depending on where in the pipeline the problem occurred. Some of the following files will not be present if they did not contain any errors.
braker/yourSpecies/errors/bam2hints.*.stderr - will give details on a bam2hints crash (step for converting bam file to intron gff file)
braker/yourSpecies/hintsfile.gff - is this file empty? If yes, something went wrong during hints generation - does this file contain hints from source “b2h” and of type “intron”? If not: GeneMark-ET will not be able to execute properly. Conversely, GeneMark-EP+ will not be able to execute correctly if hints from the source "ProtHint" are missing.
braker/yourSpecies/spaln/*err - errors reported by spaln
braker/yourSpecies/errors/GeneMark-{ET,EP,ETP}.stderr - errors reported by GeneMark-ET/EP+/ETP
braker/yourSpecies/errors/GeneMark-{ET,EP,ETP).stdout - may give clues about the point at which errors in GeneMark-ET/EP+/ETP occured
braker/yourSpecies/GeneMark-{ET,EP,ETP}/genemark.gtf - is this file empty? If yes, something went wrong during executing GeneMark-ET/EP+/ETP
braker/yourSpecies/GeneMark-{ET,EP}/genemark.f.good.gtf - is this file empty? If yes, something went wrong during filtering GeneMark-ET/EP+ genes for training AUGUSTUS
braker/yourSpecies/genbank.good.gb - try a “grep -c LOCUS genbank.good.gb” to determine the number of training genes for training AUGUSTUS, should not be low
braker/yourSpecies/errors/firstetraining.stderr - contains errors from first iteration of training AUGUSTUS
braker/yourSpecies/errors/secondetraining.stderr - contains errors from second iteration of training AUGUSTUS
braker/yourSpecies/errors/optimize_augustus.stderr - contains errors optimize_augustus.pl (additional training set for AUGUSTUS)
braker/yourSpecies/errors/augustus*.stderr - contain AUGUSTUS execution errors
braker/yourSpecies/startAlign.stderr - if you provided a protein fasta file, something went wrong during protein alignment
braker/yourSpecies/startAlign.stdout - may give clues on at which point protein alignment went wrong
BRAKER complains that the RNA-Seq file does not correspond to the provided genome file, but I am sure the files correspond to each other!
Please check the headers of the genome FASTA file. If the headers are long and contain whitespaces, some RNA-Seq alignment tools will truncate sequence names in the BAM file. This leads to an error with BRAKER. Solution: shorten/simplify FASTA headers in the genome file before running the RNA-Seq alignment and BRAKER.
GeneMark fails!
(a) GeneMark by default only uses contigs longer than 50k for training. If you have a highly fragmented assembly, this might lead to "no data" for training. You can override the default minimal length by setting the BRAKER argument --min_contig=10000 .
(b) see "[something] failed to execute" below.
[something] failed to execute!
When providing paths to software to BRAKER, please use absolute, non-abbreviated paths. For example, BRAKER might have problems with --SAMTOOLS_PATH=./samtools/ or --SAMTOOLS_PATH=~/samtools/ . Please use SAMTOOLS_PATH=/full/absolute/path/to/samtools/ , instead. This applies to all path specifications as command line options to braker.pl . Relative paths and absolute paths will not pose problems if you export a bash variable, instead, or if you append the location of tools to your $PATH variable.
GeneMark-ETP in BRAKER dies with '/scratch/11232323': No such file or directory.
This appears to be related to sorting large files, and it's a system configuration depending problem. Solve it with export TMPDIR=/tmp/ before calling BRAKER via Singularity.
BRAKER cannot find the Augustus script XYZ...
Update Augustus from github with git clone https://github.com/Gaius-Augustus/Augustus.git . Do not use Augustus from other sources. BRAKER is highly dependent on an up-to-date Augustus. Augustus releases happen rather rarely, updates to the Augustus scripts folder occur rather frequently.
Does BRAKER depend on Python3?
そうです。 The python scripts employed by BRAKER are not compatible with Python2.
Why does BRAKER predict more genes than I expected?
If transposable elements (or similar) have not been masked appropriately, AUGUSTUS tends to predict those elements as protein coding genes. This can lead to a huge number genes. You can check whether this is the case for your project by BLASTing (or DIAMONDing) the predicted protein sequences against themselves (all vs. all) and counting how many of the proteins have a high number of high quality matches. You can use the output of this analysis to divide your gene set into two groups: the protein coding genes that you want to find and the repetitive elements that were additionally predicted.
I am running BRAKER in Anaconda and something fails...
Update AUGUSTUS and BRAKER from github with git clone https://github.com/Gaius-Augustus/Augustus.git and git clone https://github.com/Gaius-Augustus/BRAKER.git . The Anaconda installation is great, but it relies on releases of AUGUSTUS and BRAKER - which are often lagging behind. Please use the current GitHub code, instead.
Why and where is the GenomeThreader support gone?
BRAKER is a joint project between teams from University of Greifswald and Georgia Tech. While the group of Mark Bordovsky from Georgia Tech contributes GeneMark expertise, the group of Mario Stanke from University of Greifswald contributes AUGUSTUS expertise. Using GenomeThreader to build training genes for AUGUSTUS in BRAKER circumvents execution of GeneMark. Thus, the GenomeThreader mode is strictly speaking not part of the BRAKER project. The previous functionality of BRAKER with GenomeThreader has been moved to GALBA at https://github.com/Gaius-Augustus/GALBA. Note that GALBA has also undergone extension for using Miniprot instead of GenomeThreader.
My BRAKER gene set has too many BUSCO duplicates!
AUGUSTUS within BRAKER can predict alternative splicing isoforms. Also the merge of the AUGUSTUS and GeneMark gene set by TSEBRA within BRAKER may result in additional isoforms for a single gene. The BUSCO duplicates usually come from alternative splicing isoforms, ie they are expected.
Augustus and/or etraining within BRAKER complain that the file aug_cmdln_parameters.json is missing. Even though I am using the latest Singularity container!
BRAKER copies the AUGUSTUS_CONFIG_PATH folder to a writable location. In older versions of Augustus, that file was indeed not existing. If the local writable copy of a folder already exists, BRAKER will not re-copy it. Simply delete the old folder. (It is often ~/.augustus , so you can simply do rm -rf ~/.augustus ; the folder might be residing in $PWD if your home directory was not writable).
I sit behind a firewall, compleasm cannot download the BUSCO files, what can I do? See Issue #785 (comment)
Since BRAKER is a pipeline that calls several Bioinformatics tools, publication of results obtained by BRAKER requires that not only BRAKER is cited, but also the tools that are called by BRAKER. BRAKER will output a file what-to-cite.txt in the BRAKER working directory, informing you about which exact sources apply to your run.
Always cite:
Stanke, M., Diekhans, M., Baertsch, R. and Haussler, D. (2008). Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics, doi: 10.1093/bioinformatics/btn013.
Stanke. M., Schöffmann, O., Morgenstern, B. and Waack, S. (2006). Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics 7, 62.
If you provided any kind of evidence for BRAKER, cite:
If you provided both short read RNA-Seq evidence and a large database of proteins, cite:
Gabriel, L., Bruna, T., Hoff, KJ, Ebel, M., Lomsadze, A., Borodovsky, M., Stanke, M. (2023). BRAKER3: Fully Automated Genome Annotation Using RNA-Seq and Protein Evidence with GeneMark-ETP, AUGUSTUS and TSEBRA. bioRxiV, doi: 10.1101/2023.06.10.54444910.1101/2023.01.01.474747.
Bruna, T., Lomsadze, A., Borodovsky, M. (2023). GeneMark-ETP: Automatic Gene Finding in Eukaryotic Genomes in Consistence with Extrinsic Data. bioRxiv, doi: 10.1101/2023.01.13.524024.
Kovaka, S., Zimin, AV, Pertea, GM, Razaghi, R., Salzberg, SL, & Pertea, M. (2019). Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome biology, 20(1):1-13.
Pertea, G., & Pertea, M. (2020). GFF utilities: GffRead and GffCompare. F1000Research, 9.
Quinlan, AR (2014). BEDTools: the Swiss‐army tool for genome feature analysis. Current protocols in bioinformatics, 47(1):11-12.
If the only source of evidence for BRAKER was a large database of protein sequences, cite:
If the only source of evidence for BRAKER was RNA-Seq data, cite:
Hoff, KJ, Lange, S., Lomsadze, A., Borodovsky, M. and Stanke, M. (2016). BRAKER1: unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics, 32(5):767-769.
Lomsadze, A., Paul DB, and Mark B. (2014) Integration of Mapped Rna-Seq Reads into Automatic Training of Eukaryotic Gene Finding Algorithm. Nucleic Acids Research 42(15): e119--e119
If you called BRAKER3 with an IsoSeq BAM file, or if you envoked the --busco_lineage option, cite:
If you called BRAKER with the --busco_lineage option, in addition, cite:
Simão, FA, Waterhouse, RM, Ioannidis, P., Kriventseva, EV, & Zdobnov, EM (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics, 31(19), 3210-3212.
Li, H. (2023). Protein-to-genome alignment with miniprot. Bioinformatics, 39(1), btad014.
Huang, N., & Li, H. (2023). compleasm: a faster and more accurate reimplementation of BUSCO. Bioinformatics, 39(10), btad595.
If any kind of AUGUSTUS training was performed by BRAKER, check carefully whether you configured BRAKER to use NCBI BLAST or DIAMOND. One of them was used to filter out redundant training gene structures.
If you used NCBI BLAST, please cite:
Altschul, AF, Gish, W., Miller, W., Myers, EW and Lipman, DJ (1990). A basic local alignment search tool. J Mol Biol 215:403--410.
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., and Madden, TL (2009). Blast+: architecture and applications. BMC bioinformatics, 10(1):421.
If you used DIAMOND, please cite:
If BRAKER was executed with a genome file and no extrinsic evidence, cite, then GeneMark-ES was used, cite:
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, YO and Borodovsky, M. (2005). Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Research, 33(20):6494--6506.
Ter-Hovhannisyan, V., Lomsadze, A., Chernoff, YO and Borodovsky, M. (2008). Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome research, pages gr--081612, 2008.
Hoff, KJ, Lomsadze, A., Borodovsky, M. and Stanke, M. (2019). Whole-Genome Annotation with BRAKER.方法mol biol。 1962:65-95, doi: 10.1007/978-1-4939-9173-0_5.
If BRAKER was run with proteins as source of evidence, please cite all tools that are used by the ProtHint pipeline to generate hints:
Bruna, T., Lomsadze, A., & Borodovsky, M. (2020). GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genomics and Bioinformatics, 2(2), lqaa026.
Buchfink, B., Xie, C., Huson, DH (2015). Fast and sensitive protein alignment using DIAMOND. Nature Methods 12:59-60.
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, YO and Borodovsky, M. (2005). Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Research, 33(20):6494--6506.
Iwata, H., and Gotoh, O. (2012). Benchmarking spliced alignment programs including Spaln2, an extended version of Spaln that incorporates additional species-specific features. Nucleic acids research, 40(20), e161-e161.
Gotoh, O., Morita, M., Nelson, DR (2014). Assessment and refinement of eukaryotic gene structure prediction with gene-structure-aware multiple protein sequence alignment. BMC bioinformatics, 15(1), 189.
If BRAKER was executed with RNA-Seq alignments in bam-format, then SAMtools was used, cite:
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R.; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25(16):2078-9.
Barnett, DW, Garrison, EK, Quinlan, AR, Strömberg, MP and Marth GT (2011). BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics, 27(12):1691-2
If BRAKER downloaded RNA-Seq libraries from SRA using their IDs, cite SRA, SRA toolkit, and HISAT2:
Leinonen, R., Sugawara, H., Shumway, M., & International Nucleotide Sequence Database Collaboration. (2010)。 The sequence read archive. Nucleic acids research, 39(suppl_1), D19-D21.
SRA Toolkit Development Team (2020). SRA Toolkit. https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software.
Kim, D., Paggi, JM, Park, C., Bennett, C., & Salzberg, SL (2019). HISAT2およびHISATジェノタイプを使用したグラフベースのゲノムアライメントとジェノタイピング。 Nature biotechnology, 37(8):907-915.
If BRAKER was executed using RNA-Seq data in FASTQ format, cite HISAT2:
If BRAKER called MakeHub for creating a track data hub for visualization of BRAKER results with the UCSC Genome Browser, cite:
If BRAKER called GUSHR for generating UTRs, cite:
Keilwagen, J., Hartung, F., Grau, J. (2019) GeMoMa: Homology-based gene prediction utilizing intron position conservation and RNA-seq data.方法mol biol。 1962:161-177, doi: 10.1007/978-1-4939-9173-0_9.
Keilwagen, J., Wenk, M., Erickson, JL, Schattat, MH, Grau, J., Hartung F. (2016) Using intron position conservation for homology-based gene prediction. Nucleic Acids Research, 44(9):e89.
Keilwagen, J., Hartung, F., Paulini, M., Twardziok, SO, Grau, J. (2018) Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinformatics, 19(1):189.
All source code, ie scripts/*.pl or scripts/*.py are under the Artistic License (see http://www.opensource.org/licenses/artistic-license.php).
[F1] EX = ES/ET/EP/ETP, all available for download under the name GeneMark-ES/ET/EP ↩
[F2] Please use the latest version from the master branch of AUGUSTUS distributed by the original developers, it is available from github at https://github.com/Gaius-Augustus/Augustus. Problems have been reported from users that tried to run BRAKER with AUGUSTUS releases maintained by third parties, ie Bioconda. ↩
[F4] install with sudo apt-get install cpanminus ↩
[F6] The binary may eg reside in bamtools/build/src/toolkit ↩
[R0] Bruna, Tomas, Hoff, Katharina J., Lomsadze, Alexandre, Stanke, Mario, and Borodovsky, Mark. 2021. “BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database." NAR Genomics and Bioinformatics 3(1):lqaa108.↩
[R1] Hoff, Katharina J, Simone Lange, Alexandre Lomsadze, Mark Borodovsky, and Mario Stanke. 2015. “BRAKER1: Unsupervised Rna-Seq-Based Genome Annotation with Genemark-et and Augustus.” Bioinformatics 32 (5). Oxford University Press: 767--69.↩
[R2] Lomsadze, Alexandre, Paul D Burns, and Mark Borodovsky. 2014. “Integration of Mapped Rna-Seq Reads into Automatic Training of Eukaryotic Gene Finding Algorithm.” Nucleic Acids Research 42 (15). Oxford University Press: e119--e119.↩
[R3] Stanke, Mario, Mark Diekhans, Robert Baertsch, and David Haussler. 2008. “Using Native and Syntenically Mapped cDNA Alignments to Improve de Novo Gene Finding.” Bioinformatics 24 (5). Oxford University Press: 637--44.↩
[R4] Stanke, Mario, Oliver Schöffmann, Burkhard Morgenstern, and Stephan Waack. 2006. “Gene Prediction in Eukaryotes with a Generalized Hidden Markov Model That Uses Hints from External Sources.” BMC Bioinformatics 7 (1). BioMed Central: 62.↩
[R5] Barnett, Derek W, Erik K Garrison, Aaron R Quinlan, Michael P Strömberg, and Gabor T Marth. 2011. “BamTools: A C++ Api and Toolkit for Analyzing and Managing Bam Files.” Bioinformatics 27 (12). Oxford University Press: 1691--2.↩
[R6] Li, Heng, Handsaker, Bob, Alec Wysoker, Tim Fennell, Jue Ruan, Nils Homer, Gabor Marth, Goncalo Abecasis, and Richard Durbin. 2009. “The Sequence Alignment/Map Format and Samtools.” Bioinformatics 25 (16). Oxford University Press: 2078--9.↩
[R7] Gremme, G. 2013. “Computational Gene Structure Prediction.” PhD thesis, Universität Hamburg.↩
[R8] Gotoh, Osamu. 2008a. “A Space-Efficient and Accurate Method for Mapping and Aligning cDNA Sequences onto Genomic Sequence.” Nucleic Acids Research 36 (8). Oxford University Press: 2630--8.↩
[R9] Iwata, Hiroaki, and Osamu Gotoh. 2012. “Benchmarking Spliced Alignment Programs Including Spaln2, an Extended Version of Spaln That Incorporates Additional Species-Specific Features.” Nucleic Acids Research 40 (20). Oxford University Press: e161--e161.↩
[R10] Osamu Gotoh. 2008b. “Direct Mapping and Alignment of Protein Sequences onto Genomic Sequence.” Bioinformatics 24 (21). Oxford University Press: 2438--44.↩
[R11] Slater, Guy St C, and Ewan Birney. 2005. “Automated Generation of Heuristics for Biological Sequence Comparison.” BMC Bioinformatics 6(1). BioMed Central: 31.↩
[R12] Altschul, SF, W. Gish, W. Miller, EW Myers, and DJ Lipman. 1990. “Basic Local Alignment Search Tool.” Journal of Molecular Biology 215:403--10.↩
[R13] Camacho, Christiam, et al. 2009. “BLAST+: architecture and applications.“ BMC Bioinformatics 1(1): 421.↩
[R14] Lomsadze, A., V. Ter-Hovhannisyan, YO Chernoff, and M. Borodovsky. 2005. “Gene identification in novel eukaryotic genomes by self-training algorithm.” Nucleic Acids Research 33 (20): 6494--6506. doi:10.1093/nar/gki937.↩
[R15] Ter-Hovhannisyan, Vardges, Alexandre Lomsadze, Yury O Chernoff, and Mark Borodovsky. 2008. “Gene Prediction in Novel Fungal Genomes Using an Ab Initio Algorithm with Unsupervised Training.” Genome Research . Cold Spring Harbor Lab, gr--081612.↩
[R16] Hoff, KJ 2019. MakeHub: Fully automated generation of UCSC Genome Browser Assembly Hubs. Genomics, Proteomics and Bioinformatics , in press, preprint on bioarXive, doi: https://doi.org/10.1101/550145.↩
[R17] Bruna, T., Lomsadze, A., & Borodovsky, M. 2020. GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genomics and Bioinformatics, 2(2), lqaa026. doi: https://doi.org/10.1093/nargab/lqaa026.↩
[R18] Kriventseva, EV, Kuznetsov, D., Tegenfeldt, F., Manni, M., Dias, R., Simão, FA, and Zdobnov, EM 2019. OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Research, 47(D1), D807-D811.↩
[R19] Keilwagen, J., Hartung, F., Grau, J. (2019) GeMoMa: Homology-based gene prediction utilizing intron position conservation and RNA-seq data.方法mol biol。 1962:161-177, doi: 10.1007/978-1-4939-9173-0_9.↩
[R20] Keilwagen, J., Wenk, M., Erickson, JL, Schattat, MH, Grau, J., Hartung F. (2016) Using intron position conservation for homology-based gene prediction. Nucleic Acids Research, 44(9):e89.↩
[R21] Keilwagen, J., Hartung, F., Paulini, M., Twardziok, SO, Grau, J. (2018) Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinformatics, 19(1):189.↩
[R22] SRA Toolkit Development Team (2020). SRA Toolkit. https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software.[↩](#a22)
[R23] Kim, D., Paggi, JM, Park, C., Bennett, C., & Salzberg, SL (2019). HISAT2およびHISATジェノタイプを使用したグラフベースのゲノムアライメントとジェノタイピング。 Nature biotechnology, 37(8):907-915.↩
[R24] Quinlan, AR (2014). BEDTools: the Swiss‐army tool for genome feature analysis. Current protocols in bioinformatics, 47(1):11-12.↩
[R25] Kovaka, S., Zimin, AV, Pertea, GM, Razaghi, R., Salzberg, SL, & Pertea, M. (2019). Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome biology, 20(1):1-13.↩
[R26] Pertea, G., & Pertea, M. (2020). GFF utilities: GffRead and GffCompare. F1000Research, 9.↩
[R27] Huang, N., & Li, H. (2023). compleasm: a faster and more accurate reimplementation of BUSCO. Bioinformatics, 39(10), btad595.↩
[R28] Bruna, T., Gabriel, L. & Hoff, KJ (2024). Navigating Eukaryotic Genome Annotation Pipelines: A Route Map to BRAKER, Galba, and TSEBRA. arXiv, https://doi.org/10.48550/arXiv.2403.19416 .↩


