ClockstaR
1.0.0

Sebastian Duchene, Martyna Molak e Simon YW Ho.
Laboratório de Ecologia Molecular, Evolução e Filogenética (MEEP)
Escola de Ciências Biológicas
Universidade de Sydney
10 de junho de 2015
partition_data_partitionfinder('drag fasta file with concatenated data here', 'drag partition finder output here')
optim.trees.interactive(folder.parts = 'path to your folder with fasta files and tree topology here')
Implementar otimização de distâncias de árvores usando a derivada da distância BSD
Implementar versão paralela para distância de topologia
Escreva tutorial para clustering de distância de topologia
Integrar modelgenerator para teste de modelo
Integre o RaxML para otimização de máxima probabilidade de comprimentos de ramificação e topologias
Estimar escalas de tempo evolutivas com conjuntos de dados multigênicos é um exercício comum em estudos filogenéticos. Os conjuntos de dados multigênicos podem ser particionados por gene, posição de códon ou ambos. Neste tutorial, nos referimos a “subconjuntos de dados” como genes individuais ou qualquer subunidade do conjunto de dados multigênicos. O termo “partições” se referirá a um grupo de subconjuntos de dados.
Embora os subconjuntos de dados possam ser concatenados e analisados com um único modelo de relógio relaxado, os padrões de variação da taxa entre linhagens podem diferir entre os subconjuntos de dados, mesmo quando suas topologias de árvore são idênticas. Por exemplo, a variação da taxa entre linhagens nos genes mitocondriais pode diferir daquela dos genes nucleares. Portanto, diferentes modelos de relógio relaxado podem ser atribuídos a diferentes subconjuntos de dados, a fim de melhorar as estimativas de escalas de tempo evolutivas e ajuste estatístico (ver Duchene e Ho., 2014a).
Há um grande número de maneiras pelas quais conjuntos de dados multigênicos podem ser particionados. Uma abordagem comum para comparar esquemas de particionamento é usar fatores de Bayes ou critérios baseados em probabilidade para ajuste do modelo. Na maioria dos casos, entretanto, é inviável testar todos os esquemas de particionamento possíveis, especialmente com métodos computacionalmente intensivos de cálculo dos fatores de Bayes.
ClockstaR estima os comprimentos dos ramos filogenéticos de cada subconjunto de dados. A distância da pontuação do ramo, conhecida como sBSDmin, é calculada para cada par de árvores como uma medida da diferença em seus padrões de variação da taxa entre linhagens. Essas distâncias são usadas para inferir a melhor estratégia de particionamento usando a estatística GAP com o algoritmo de clustering PAM, conforme implementado no cluster de pacotes (Maechler et al., 2012) (para detalhes da métrica sBSDmin, consulte Duchene et al., 2014b) .
ClockstaR é um pacote R para análises filogenéticas de relógio molecular de conjuntos de dados multigênicos. Ele usa os padrões de variação da taxa de linhagem para os diferentes genes para selecionar a estratégia de particionamento do relógio. O método usa uma métrica de distância de árvore filogenética e um algoritmo de aprendizado de máquina não supervisionado para identificar o número ideal de partições de relógio e quais genes devem ser analisados em cada uma das partições. A estratégia de particionamento selecionada no ClocsktaR pode ser usada para posterior análise do relógio molecular com programas como BEAST, MrBayes, PhyloBayes e outros.
Por favor, siga este link para a publicação original.
ClockstaR requer uma instalação R. Também requer algumas dependências do R, que podem ser obtidas através do R, conforme explicado a seguir.
Por favor, envie quaisquer solicitações ou perguntas para Sebastian Duchene (sebastian.duchene[at]sydney.edu.au). Alguns outros softwares e recursos podem ser encontrados no Laboratório de Ecologia Molecular, Evolução e Filogenética da Universidade de Sydney.
Baixe este repositório como um arquivo zip e descompacte-o. As instruções a seguir usam a pasta clockstar_example_data, que contém alguns arquivos fasta e uma árvore filogenética no formato newick. Abra qualquer um desses arquivos em um editor de texto, como o text wrangler. Esses dados foram simulados sob quatro padrões de variação da taxa evolutiva. Observe que a árvore é a topologia em árvore para todos os genes ou partições de dados. Para executar o ClockstaR, formate seus dados de maneira semelhante aos dados de exemplo em clockstar_example_data.
ClockstaR pode ser instalado diretamente do GitHub. Isso requer o pacote devtools. Digite o seguinte código no prompt R para instalar todas as ferramentas necessárias (observe que você precisará de conexão com a Internet para baixar os pacotes diretamente):
install . packages ( " devtools " )
library (devtools)
install_github ( ' ClockstaR ' , ' sebastianduchene ' )Após baixar e instalar, carregue o ClockstaR com a biblioteca de funções.
library (ClockstaR2)Para ver um exemplo de como o programa é executado digite:
example (ClockstaR2)O restante deste tutorial usa a pasta clockstar_example_data
O primeiro passo é obter as árvores genéticas para cada um dos alinhamentos. Para fazer isso, usamos a topologia de árvore e otimizamos os comprimentos dos ramos usando cada um dos alinhamentos de genes individuais, neste caso A1.fasta até C3.fasta. Se você tiver as árvores genéticas, salve-as no formato newick em um arquivo e passe para a próxima etapa (executar o clockstar interativamente).
Digite o seguinte código no prompt R e pressione Enter:
optim . trees . interactive ()Se você receber uma mensagem de erro sobre a instalação do pacote phangorn, use este código e repita optim.trees.interactive()
install . packcages ( " phangorn " )ClockstaR imprimirá a seguinte mensagem:
Please drag a folder with the data subsets and a tree topology . The files should be in FASTA format, and the trees in NEWICKArraste a pasta clockstar_example_data para o console R e digite enter. Observe que a pasta deve conter apenas os alinhamentos no formato FASTA e a topologia em árvore no NEWICK. Você verá a seguinte mensagem:
What should be the name of the file to save the optimised trees ?Digite o nome do arquivo para as árvores otimizadas. Neste caso usaremos "example.trees"
example . treesNeste ponto, ClockstaR perguntará se deve usar um modelo de substituição separado para cada gene ou usar JC em todos os casos. Como esses dados foram simulados no JC, digitaremos “n” e pressionaremos enter. Digite "y" para especificar cada modelo de substituição separadamente.
Após digitar “n” e pressionar enter, o ClockstaR começará a funcionar. Ele imprimirá as árvores genéticas no dispositivo gráfico. Se a árvore especificada estiver enraizada, ela também poderá imprimir alguns avisos, que podem ser ignorados com segurança.
Abra a pasta clockstar_example_data. Você encontrará um arquivo com o nome "example.trees", conforme especificado alguns passos acima. Abra example.trees em um editor de texto. Ele contém cada árvore genética e os nomes das árvores, de acordo com os nomes dos alinhamentos genéticos. Deve ser algo assim:
A1 . fasta (( t1 : 0.01504695462 ,( t2 : 0.00987 ...
A2 . fasta (( t1 : 0.01520523401 ,( t2 : 0.01317 ...
A3 . fasta (( t1 : 0.01519309467 ,( t2 : 0.01092 ...
.
.
.Este arquivo com árvores será utilizado na próxima etapa.
Para esta etapa é necessário ter as árvores genéticas em um arquivo, como o obtido na etapa anterior.
Abra R e carregue ClockstaR conforme mostrado acima. Digite o seguinte código no prompt:
clockstar . interactive ()ClockstaR imprimirá a seguinte mensagem:
please drag or type in the path to your gene trees file in NEWICK format :Arraste o arquivo com as árvores genéticas para o console R. Se você seguiu a etapa anterior, o arquivo se chamará example.trees. Digite entrar.
Dependendo dos pacotes que você instalou, o ClockstaR pode perguntar se deve rodar em paralelo. Isso é eficiente para grandes conjuntos de dados. Mas para os dados de exemplo não fará muita diferença, então digite "n" se vir esta mensagem e depois digite enter:
Packages foreach and doParallel are available for parallel computation
Should we run ClockstaR in parallel (y / n) ? (This is good for large data sets)Clockstar agora começará a funcionar. A saída na tela deve ser semelhante a esta:
[ 1 ] " Calculating sBSDmin distances between all pairs of trees "
[ 1 ] " Estimating tree distances "
[ 1 ] " estimating distances 1 of 11 "
[ 1 ] " estimating distances 2 of 11 "
[ 1 ] " estimating distances 3 of 11 "
[ 1 ] " estimating distances 4 of 11 "
[ 1 ] " estimating distances 5 of 11 "
.
.
.Após estimar as distâncias das árvores (descritas na publicação original), ClockstaR imprimirá a seguinte mensagem:
" I finished calculating the sBSDmin distances between trees "
The settings for clustering with ClockstaR are :
PAM clustering algorithm
K from 1 to number of data subsets - 1
SEmax criterion to select the optimal k
500 bootstrap replicates
Are these correct ? (y / n)Estas são as configurações do algoritmo de cluster. Eles são apropriados para a maioria dos conjuntos de dados, portanto neste exemplo podemos digitar “y” e depois enter. Ao digitar “n” podemos alterar essas configurações, para mais detalhes veja Kaufman e Rousseeuw (2009).
ClockstaR agora executará o algoritmo de cluster. Ao final imprimirá o melhor número de partições e perguntará se os resultados devem ser salvos em arquivo pdf:
[ 1 ] " ClockstaR has finished running "
[ 1 ] " The best number of partitions for your data set is: 3 "
Do you wish to save the results in a pdf file ? (y / n)Digite "y" e entre.
ClockstaR irá então pedir o nome dos arquivos de saída:
What should be the name and path of the output file ?Para este exemplo digite "example_run" e digite, mas qualquer nome pode ser usado.
Agora abra a pasta clockstar_example_data e abra os dois arquivos pdf, example_run_gapstats.pdf e example_run_matrix.pdf.
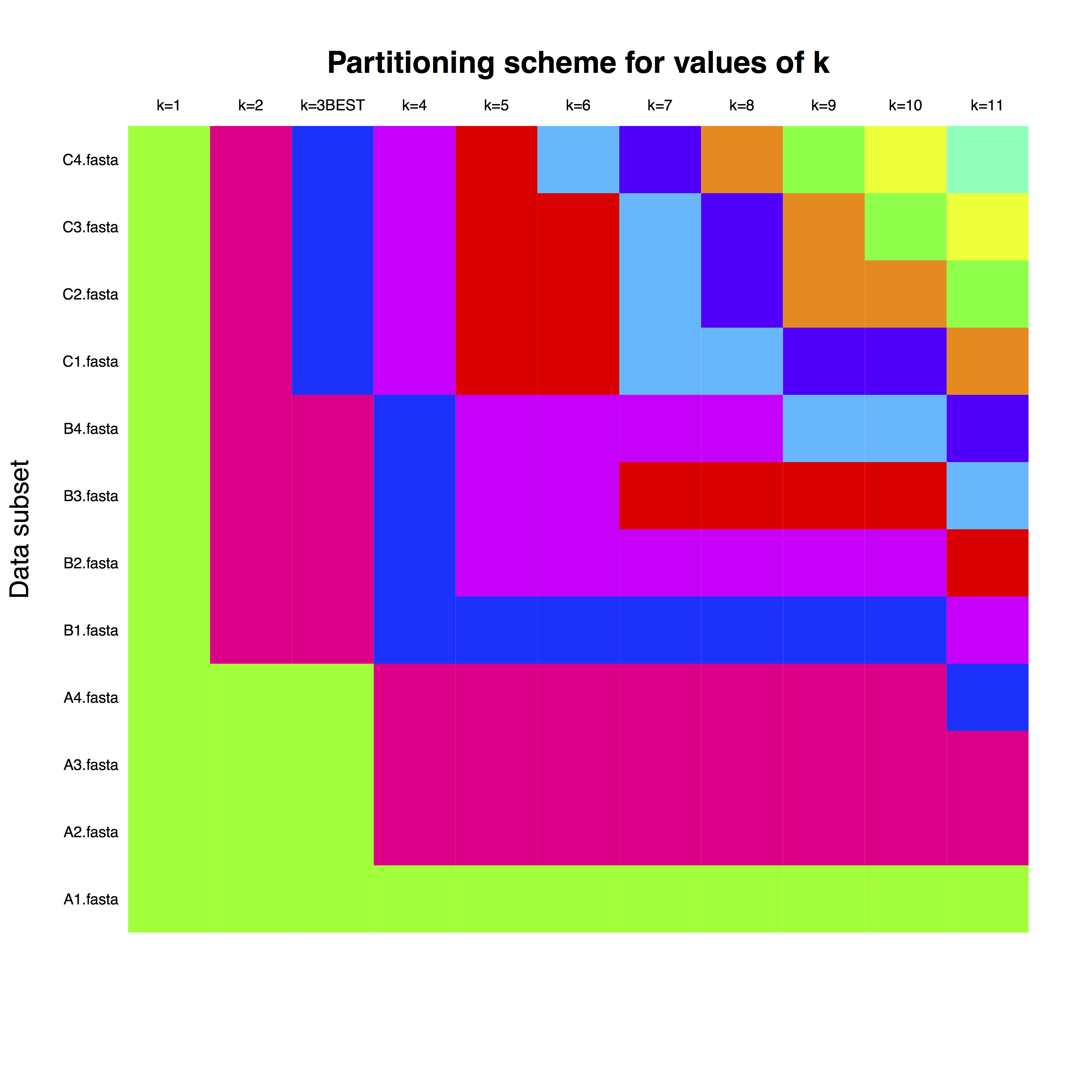
example_run_matrix é uma matriz, onde as linhas correspondem a cada gene, conforme nomeado nos arquivos FASTA. As colunas representam o número de partições e as cores representam a atribuição de cada gene à partição do relógio. Por exemplo, para k =3, que é o melhor número de partições, pode-se usar partições de relógio separadas para genes com letras A, B e C.
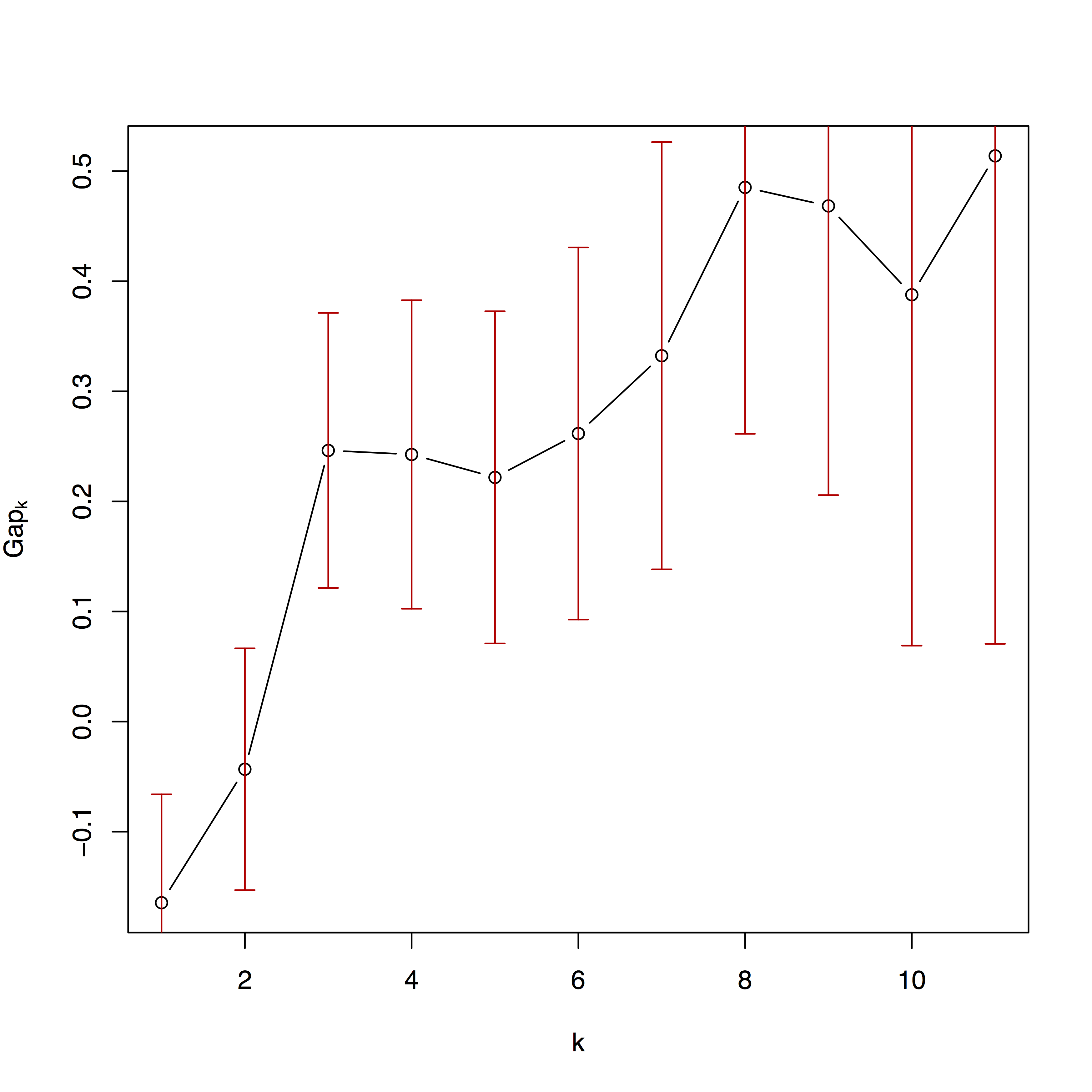
O segundo gráfico é o ajuste dos algoritmos de agrupamento em diferentes números de partições. Mais detalhes estão disponíveis em Kaufman e Rousseeuw (2009) e na documentação para cluster de pacotes.
ClockstaR pode ser executado com outras configurações personalizadas. Consulte a documentação para obter outros detalhes ou envie-me um e-mail para qualquer dúvida em sebastian.duchene[at]sydney.edy.au.
O logotipo foi desenhado por Jun Tong
Duchene, S. e Ho, SY (2014a). Usando vários modelos de relógio relaxado para estimar escalas de tempo evolutivas a partir de dados de sequência de DNA. Filogenética Molecular e Evolução (77): 65-70.
Duchene, S., Molak, M., & Ho, SY (2014b). ClockstaR: escolha do número de modelos de relógio relaxado na análise filogenética molecular. Bioinformática 30 (7): 1017-1019.
Kaufman, L. e Rousseeuw, PJ (2009). Encontrando grupos em dados: uma introdução à análise de cluster (Vol. 344). John Wiley e Filhos.


