Este repositório contém:
sepal requer python3 , de preferência uma versão posterior ou igual a 3.5. Para baixar e instalar, abra o terminal e mude para um diretório onde deseja que sepal seja baixado e faça:
git clone https://github.com/almaan/sepal.git
cd sepal
chmod +x setup.py
./setup.py install
Dependendo dos seus privilégios de usuário, pode ser necessário adicionar --user como argumento para setup.py . A execução da configuração fornecerá a instalação mínima necessária para calcular os tempos de difusão. Porém, se quiser usar os módulos de análise, você também precisará instalar os pacotes recomendados. Para fazer isso, simplesmente (no mesmo diretório) execute:
pip install -e " .[full] " novamente, o --user pode ser necessário incluir. Além disso, pode ser necessário usar pip3 se for assim que você configurou sua interface python-pip . Se você estiver usando ambientes conda ou virtuais, siga as recomendações para instalação de pacotes.
Isso deve instalar uma interface de linha de comando (CLI) e um pacote padrão. Para testar e ver se a instalação foi bem sucedida você pode tentar executar o comando:
sepal -h
Que deve imprimir a mensagem de ajuda associada ao sépala. Se tudo deu certo para você até agora, você pode prosseguir para a seção de exemplos para ver sepal em ação!
O uso recomendado de sépala é pela interface de linha de comando. Tanto as simulações para calcular os tempos de difusão quanto a posterior análise ou inspeção dos resultados podem ser facilmente realizadas digitando sepal seguido de run ou analyze . O módulo analyze possui diferentes opções, para visualizar os resultados ( inspect ), ordenar os perfis em famílias de padrões ( family ) ou submeter as famílias identificadas à análise de enriquecimento funcional ( fea ). Para obter uma lista completa de comandos disponíveis, faça sepal module -h , onde module é run e analyze . Abaixo, ilustramos como a sépala pode ser usada para encontrar perfis de transcrição com padrões espaciais.
Criaremos uma pasta para armazenar nossos resultados, que também servirá como nosso diretório de trabalho. No diretório principal do repositório, faça:
cd res
mkdir example
cd exampleA amostra MOB será utilizada para exemplificar nossa análise. Começamos calculando os tempos de difusão para cada perfil de transcrição:
sepal run -c ../../data/real/mob.tsv.gz -mo 10 -mc 5 -o . -ar 1Abaixo está um exemplo (com uma exibição adicional do comando de ajuda) de como isso pode parecer

Tendo calculado os tempos de difusão, queremos inspecionar o resultado, como no estudo, veremos os 20 principais perfis. Podemos facilmente gerar imagens a partir do nosso resultado executando o comando:
sepal analyze -c ../../data/real/mob.tsv.gz
-r 20200409173043610345-top-diffusion-times.tsv
-ar 1k -o . inspect -ng 20 -nc 5O que seria algo parecido com isto:

A saída será a seguinte imagem:
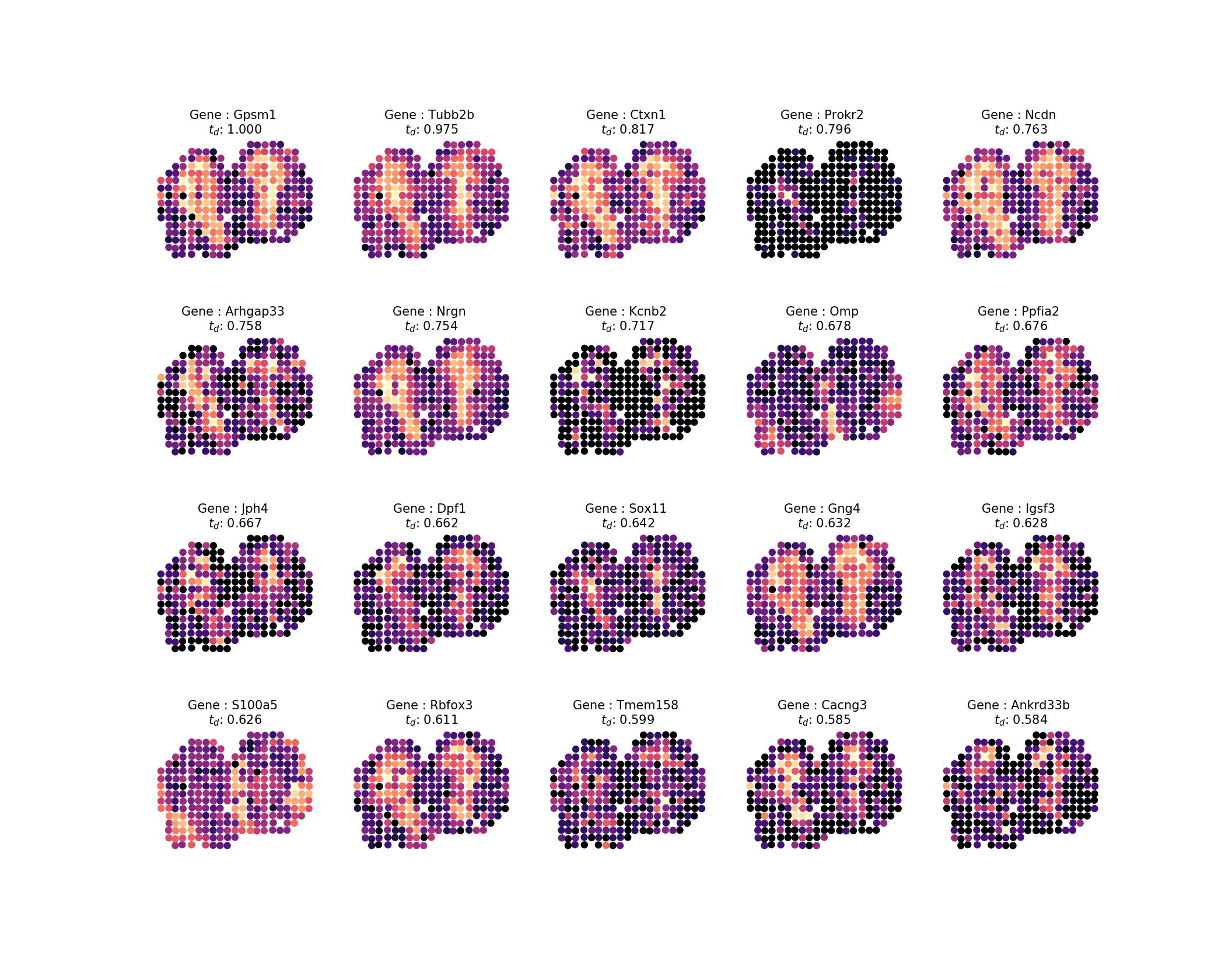
Então, para classificar os 100 genes mais bem classificados em um conjunto de famílias de padrões, onde 85% da variação em nossos padrões deveria ser explicada pelos padrões próprios, faça:
sepal analyze -c ../../data/real/mob.tsv.gz
-r 20200409173043610345-top-diffusion-times.tsv
-ar 1k -o . family -ng 100 -nbg 100 -eps 0.85 --plot -nc 3Disto obtemos os seguintes três motivos representativos de cada família:
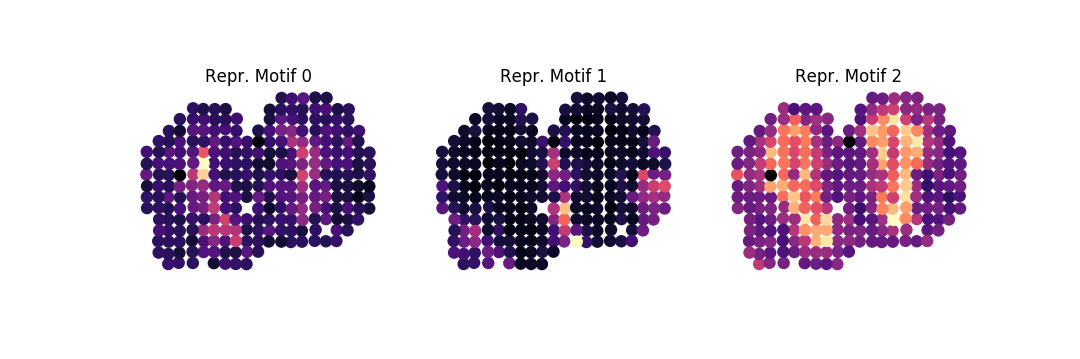
Podemos submeter nossas famílias a análises de enriquecimento, executando:
sepal analyze -c ../../data/real/mob.tsv.gz
-r 20200409173043610345-top-diffusion-times.tsv
-ar 1k -o . fea -fl mob.tsv-family-index.tsv -or " mmusculus "onde vemos, por exemplo, que a Família 2 é enriquecida para vários processos relacionados à função, geração e regulação neuronal:
| família | nativo | nome | valor_p | fonte | tamanho_intersecção | |
|---|---|---|---|---|---|---|
| 2 | 2 | IR:0007399 | desenvolvimento do sistema nervoso | 0,00035977 | IR:BP | 26 |
| 3 | 2 | IR:0050773 | regulação do desenvolvimento de dendritos | 0,000835883 | IR:BP | 8 |
| 4 | 2 | IR:0048167 | regulação da plasticidade sináptica | 0,00196494 | IR:BP | 8 |
| 5 | 2 | IR:0016358 | desenvolvimento de dendritos | 0,00217167 | IR:BP | 9 |
| 6 | 2 | IR:0048813 | morfogênese dendrítica | 0,00741589 | IR:BP | 7 |
| 7 | 2 | IR:0048814 | regulação da morfogênese dendrítica | 0,00800399 | IR:BP | 6 |
| 8 | 2 | IR:0048666 | desenvolvimento de neurônios | 0,0114088 | IR:BP | 16 |
| 9 | 2 | IR:0099004 | via de sinalização de quinase dependente de calmodulina | 0,0159572 | IR:BP | 3 |
| 10 | 2 | IR:0050804 | modulação da transmissão sináptica química | 0,0341913 | IR:BP | 10 |
| 11 | 2 | IR:0099177 | regulação da sinalização transsináptica | 0,0347783 | IR:BP | 10 |
É claro que esta análise não é de forma alguma exaustiva. Mas sim um exemplo rápido para mostrar como operar a CLI para sepal .
Embora sepal tenha sido projetado como uma ferramenta independente, também o construímos para funcionar como um pacote python padrão a partir do qual as funções podem ser importadas e usadas em um fluxo de trabalho integrado. Para mostrar como isso pode ser feito, fornecemos um exemplo, reproduzindo a análise do melanoma. Mais exemplos podem ser adicionados posteriormente.
A entrada para sepal deve estar no formato n_locations x n_genes , no entanto, se seus dados estiverem estruturados de maneira oposta ( n_genes x n_locations ), basta fornecer o sinalizador --transpose ao executar a simulação ou análise e isso será resolvido de.
Atualmente oferecemos suporte aos formatos .csv , .tsv e .h5ad . Para este último, seu arquivo deverá ser estruturado de acordo com ESTE formato. Esperamos que em um futuro próximo haja um lançamento da equipe scanpy , onde seja apresentado um formato padronizado para dados espaciais, mas até então estaremos utilizando o padrão mencionado.
Todos os dados reais que utilizamos são públicos e podem ser acessados nos seguintes links:
Os dados sintéticos foram gerados por:
synthetic/img2cnt.pysynthetic/turing.pysynthetic/ablation.py Todos os resultados apresentados no estudo podem ser encontrados na pasta res , tanto para os dados reais quanto para os sintéticos. Para cada amostra estruturamos os resultados de acordo:
res/sample-name/X-diffusion-times.tsv : tempos de difusão para todos os genes classificadosanalysis/ : contém saída da análise secundária

