reference_database_creator
bug fix --in-silico-pcr --untrimmed
CARANGUEJOS ( C comendo R bancos de dados de referência para UM mplicon- B ased S equencing) é um programa de software versátil que gera bancos de dados de referência selecionados para análise metagenômica. O fluxo de trabalho do CRABS consiste em sete módulos: (i) download de dados de repositórios online; (ii) importar dados baixados para o formato CRABS; (iii) extrair regiões amplificadas através de análise de PCR in silico ; (iv) recuperar amplicons sem regiões de ligação a primers através de alinhamentos com códigos de barras extraídos in silico ; (v) organizar e criar subconjuntos do banco de dados local por meio de vários parâmetros de filtragem; (vi) exportar a base de dados local em diversos formatos de acordo com os requisitos do classificador taxonômico; e (vi) funções de pós-processamento, ou seja, visualizações, para explorar e fornecer uma visão geral resumida da base de dados de referência local. Esses sete módulos estão divididos em dezoito funções e são descritos a seguir. Além disso, é fornecido um código de exemplo para cada uma das dezoito funções. Finalmente, um tutorial para construir um banco de dados local de referência de tubarões para o conjunto de primers MiFish-E é fornecido no final deste documento README para fornecer um exemplo de script para referência.
Temos o prazer de anunciar que o CRABS passou por uma grande atualização e reformulação do código com base no feedback do usuário, o que esperamos que melhore a experiência do usuário na construção de seu próprio banco de dados de referência local!
Veja abaixo uma lista de recursos e melhorias adicionadas ao CRABS v 1.0.0 :
CRABS v 1.0.0 agora pode ser baixado manualmente clonando este repositório GitHub (consulte 4.1 Instalação manual para informações detalhadas). Atualizaremos o contêiner Docker e o pacote conda o mais rápido possível para facilitar a instalação da versão mais recente.
Ao usar CRABS em seus projetos de pesquisa, cite o seguinte artigo:
[Jeunen, G.-J., Dowle, E., Edgecombe, J., von Ammon, U., Gemmell, N. J., & Cross, H. (2022). crabs—A software program to generate curated reference databases for metabarcoding sequencing data. Molecular Ecology Resources, 00, 1– 14.](https://doi.org/10.1111/1755-0998.13741)
CRABS é um kit de ferramentas somente de linha de comando executado em ambientes Unix/Linux típicos e é escrito exclusivamente em python3. No entanto, CRABS faz uso do módulo subprocess em python para executar vários comandos na sintaxe bash para contornar idiossincrasias específicas do python e aumentar a velocidade de execução. Fornecemos três maneiras de instalar o CRABS. Para a versão mais atualizada do CRABS, recomendamos a instalação manual clonando este repositório GitHub e instalando 10 dependências separadamente (instruções de instalação para todas as dependências fornecidas em 4.1 Instalação manual). CRABS também pode ser instalado via Docker e conda. Ambos os métodos permitem uma instalação fácil, co-instalando automaticamente todas as dependências. Nosso objetivo é manter o contêiner Docker e o pacote conda atualizados, embora possa ocorrer um certo atraso na atualização para a versão mais recente, especialmente para o pacote conda. Abaixo estão os detalhes de todas as três abordagens.
Para a instalação manual, primeiro clone o repositório CRABS. Esta etapa requer que o GitHub esteja disponível na linha de comando (instruções de instalação do GitHub).
git clone https://github.com/gjeunen/reference_database_creator.git
Dependendo de suas configurações, pode ser necessário que o CRABS se torne executável em seu sistema. Isso pode ser conseguido usando o código abaixo.
chmod +x reference_database_creator/crabs
Depois que o CRABS estiver instalado, precisamos garantir que todas as dependências estejam instaladas e acessíveis globalmente. A versão mais recente do CRABS (versão v 1.0.0 ) roda em Python 3.11.7 (ou qualquer versão compatível com 3.11.7) e depende de cinco módulos Python que podem não ser padrão com Python, bem como cinco programas de software externos. Todas as dependências estão listadas abaixo, juntamente com um link para as instruções de instalação. Os números de versão fornecidos para cada módulo e programa de software são aqueles nos quais o CRABS foi desenvolvido. Embora versões compatíveis de cada um também possam ser usadas.
Módulos Python:
Programas de software externos:
Depois que o CRABS e todas as dependências estiverem instalados, o CRABS pode ficar acessível em todo o sistema operacional usando o código abaixo.
export PATH="/path/to/crabs/folder:$PATH"
Substitua /path/to/crabs/folder pelo caminho real para a pasta do repositório GitHub no sistema operacional, ou seja, a pasta criada durante o comando git clone acima. Adicionar o código export ao arquivo .bash_profile ou .bashrc tornará o CRABS acessível globalmente a qualquer momento.
Docker é um projeto de código aberto que permite a implantação de aplicativos de software dentro de 'contêineres' isolados do seu computador e executados por meio de um sistema operacional host virtual chamado Docker Engine. A principal vantagem de executar o docker em máquinas virtuais é que elas usam muito menos recursos. Esse isolamento significa que você pode executar um contêiner Docker na maioria dos sistemas operacionais, incluindo Mac, Windows e Linux. Pode ser necessário configurar uma conta gratuita para usar o Docker Desktop. Este link traz uma boa introdução aos fundamentos do uso do Docker. Aqui está um link para você começar e se orientar sobre o multiverso Docker.
Existem apenas duas etapas para fazer o Crabs funcionar no seu computador. Primeiro, instale o Docker Desktop no seu computador, que é gratuito para a maioria dos usuários. Aqui estão as instruções para Mac ; aqui estão as instruções para computadores Windows e aqui estão as instruções para Linux (a maioria das principais plataformas Linux são suportadas). Depois de instalar e executar o Docker Desktop (o aplicativo Desktop deve estar em execução para que você possa usar qualquer comando do docker na linha de comando), basta 'puxar' nossa imagem Crabs e você estará pronto para prosseguir:
docker pull quay.io/swordfish/crabs:0.1.7
Embora a instalação de um aplicativo docker seja fácil, usar esses aplicativos pode ser um pouco complicado no início. Para ajudá-lo a começar, fornecemos alguns exemplos de comandos usando a versão docker do crabs. Esses exemplos podem ser encontrados na pasta docker_intro deste repositório . A partir desses exemplos, você poderá executar a configuração de um banco de dados de referência inteiro e estar pronto para começar. Continuaremos a expandir esses exemplos e a testá-los em muitas situações diferentes. Faça perguntas e forneça comentários na guia Problemas.
Para instalar o pacote conda, você deve primeiro instalar o conda. Veja este link para detalhes. Se o conda já estiver instalado, é uma boa prática atualizar a ferramenta conda com conda update conda antes de instalar o CRABS.
Depois que o conda estiver instalado, siga as etapas abaixo para instalar o CRABS e todas as dependências. Certifique-se de inserir os comandos na ordem em que aparecem abaixo.
conda create -n CRABS
conda activate CRABS
conda config --add channels bioconda
conda config --add channels conda-forge
conda install -c bioconda crabs
Depois de inserir o comando de instalação, o conda processará a solicitação (isso pode levar um minuto ou mais) e, em seguida, exibirá todos os pacotes e programas que serão instalados e solicitará a confirmação. Digite y para iniciar a instalação. Depois que isso terminar, os CARANGUEJOS deverão estar prontos para uso.
Testamos esta instalação em sistemas Mac e Linux. Ainda não testamos no Windows Subsystem for Linux (WSL).
Use o código abaixo para verificar se o CRABS foi instalado com sucesso e obtenha as informações de ajuda.
crabs -hAs informações de ajuda dividem as dezoito funções em grupos diferentes, com cada grupo listando a função na parte superior e os parâmetros obrigatórios e opcionais abaixo.

CRABS contém sete módulos, que incorporam dezoito funções:
Módulo 1: baixar dados de repositórios online
--download-taxonomy : baixa informações de taxonomia do NCBI;--download-bold : baixa dados de sequência do banco de dados Barcode of Life (BOLD);--download-embl : baixa dados de sequência do European Nucleotide Archive (ENA; EMBL);--download-mitofish : baixa dados de sequência do banco de dados MitoFish;--download-ncbi : baixa dados de sequência do Centro Nacional de Informações sobre Biotecnologia (NCBI).Módulo 2: importar dados baixados para o formato CRABS
--import : importa sequências baixadas ou códigos de barras personalizados para o formato CRABS;--merge : mescla diferentes arquivos formatados em CRABS em um único arquivo.Módulo 3: extrair regiões amplificadas por meio de análise de PCR in silico
--in-silico-pcr : extrai amplicons de dados baixados, localizando e removendo regiões de ligação de primer.Módulo 4: recuperar amplicons sem regiões de ligação ao primer
--pairwise-global-alignment : recupera amplicons sem regiões de ligação de primer, alinhando sequências baixadas com códigos de barras extraídos in silico .Módulo 5: curadoria e subdivisão do banco de dados local por meio de vários parâmetros de filtragem
--dereplicate : descarta sequências duplicadas;--filter : descarta sequências por meio de vários parâmetros de filtragem;--subset : subconjunto do banco de dados local para reter ou excluir grupos taxonômicos especificados.Módulo 6: exportar o banco de dados local
--export : exporta o banco de dados formatado em CRABS para vários formatos de acordo com os requisitos do classificador taxonômico a ser usado.Módulo 7: funções de pós-processamento para explorar e fornecer uma visão geral resumida do banco de dados de referência local
--diversity-figure : cria um gráfico de barras horizontais exibindo o número de espécies e grupos de sequências por nível especificado incluídos na base de dados de referência;--amplicon-length-figure : cria um gráfico de linhas representando distribuições de comprimento de amplicon separadas por grupo taxonômico;--phylogenetic-tree : cria uma árvore filogenética com códigos de barras do banco de dados de referência para uma lista alvo de espécies;--amplification-efficiency-figure : cria um gráfico de barras exibindo incompatibilidades nas regiões de ligação do primer;--completeness-table : cria uma planilha contendo a disponibilidade de códigos de barras para grupos taxonômicos.Os dados de sequenciamento inicial podem ser baixados pelo CRABS de quatro repositórios online, incluindo (i) BOLD, (ii) EMBL, (iii) MitoFish e NCBI. A partir da versão v 1.0.0 , o download dos dados de cada repositório é dividido em funções próprias. Além disso, o CRABS não formata automaticamente os dados após o download para aumentar a flexibilidade e permitir a depuração quando o download dos dados falha.
Além de baixar dados de sequência, o CRABS também é capaz de baixar as informações de taxonomia do NCBI, que o CRABS usa para criar a linhagem taxonômica para cada sequência.
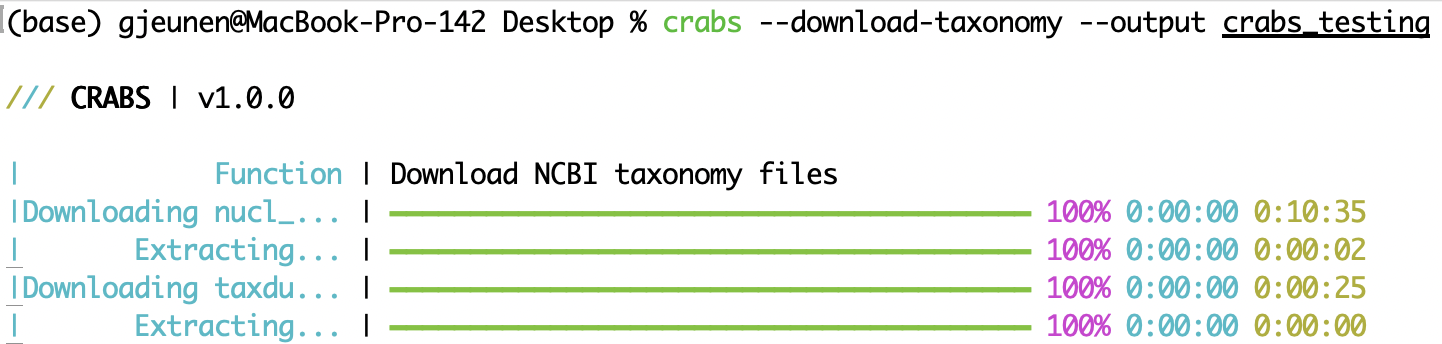
--download-taxonomy Para atribuir uma linhagem taxonômica a cada sequência baixada no banco de dados de referência (ver 5.2 Módulo 2), a informação taxonômica precisa ser baixada. CRABS utiliza a taxonomia do NCBI e baixa três arquivos específicos para o seu computador: (i) um arquivo ligando números de acesso a IDs taxonômicos ( nucl_gb.accession2taxid ), (ii) um arquivo contendo informações sobre o nome filogenético associado a cada ID taxonômico ( names.dmp ) e (iii) um arquivo contendo informações sobre como os IDs taxonômicos estão vinculados ( nós.dmp ). O diretório de saída dos arquivos baixados pode ser especificado usando o parâmetro --output . Para excluir o arquivo nucl_gb.accession2taxid ou os arquivos names.dmp e nodes.dmp , o parâmetro --exclude acc2tax ou --exclude taxdump pode ser fornecido, respectivamente. O primeiro código abaixo não baixa nenhum arquivo, pois tanto acc2tax quanto taxdump são fornecidos para o parâmetro --exclude . A segunda linha de código baixa todos os três arquivos para o subdiretório --output crabs_testing . A captura de tela abaixo mostra o que é impresso no console ao executar esta linha de código.
crabs --download-taxonomy --exclude 'acc2taxid,taxdump'
crabs --download-taxonomy --output crabs_testing
--download-bold As sequências BOLD são baixadas através do site BOLD. O arquivo de saída, que é estruturado como um documento fasta de duas linhas, pode ser especificado usando o parâmetro --output . Os usuários podem especificar qual grupo taxonômico baixar usando o parâmetro --taxon . Recomendamos escrever um loop for simples (exemplo fornecido abaixo) quando os usuários quiserem baixar vários grupos taxonômicos, limitando assim a quantidade de dados a serem baixados do BOLD por instância. No entanto, se apenas um número limitado de grupos taxonômicos for de interesse, os nomes dos grupos taxonômicos também poderão ser separados por | (exemplo fornecido abaixo). Também recomendamos que os usuários verifiquem se o nome do grupo taxonômico a ser baixado está listado no arquivo BOLD ou se nomes alternativos precisam ser usados. Por exemplo, especificar --taxon Chondrichthyes não fará o download de todas as sequências de peixes cartilaginosos do BOLD, uma vez que o nome desta classe não está listado no BOLD. Os usuários devem usar --taxon Elasmobranchii neste caso. Os usuários também podem especificar o limite do download para um marcador genético específico, fornecendo o parâmetro --marker . Quando vários marcadores genéticos são de interesse, os nomes dos marcadores devem ser separados por | . Os quatro principais marcadores de código de barras de DNA no BOLD são COI-5P , ITS , matK e rbcL . A entrada para o parâmetro --marker diferencia maiúsculas de minúsculas.
Abordagem recomendada: Um loop for simples para baixar dados do BOLD para vários grupos taxonômicos (abordagem recomendada). O código abaixo primeiro baixa os dados de Elasmobranchii, seguidos pelas sequências atribuídas a Mammalia. Os dados baixados serão gravados no subdiretório --output crabs_testing e colocados em dois arquivos separados, indicando quais dados pertencem a qual grupo taxonômico, ou seja, crabs_testing/bold_Elasmobranchii.fasta e crabs_testing/bold_Mammalia.fasta .
for taxon in Elasmobranchii Mammalia; do crabs --download-bold --taxon ${taxon} --output crabs_testing/bold_${taxon}.fasta; done

Opção alternativa: Além do loop for recomendado, vários nomes de táxons podem ser fornecidos de uma vez, separando os nomes usando | .
crabs --download-bold --taxon 'Elasmobranchii|Mammalia' --output crabs_testing/bold_elasmobranchii_mammalia.fasta
--download-embl As sequências do EMBL são baixadas através do site FTP da ENA. Os arquivos EMBL serão baixados primeiro no formato '.fasta.gz' e serão descompactados automaticamente assim que o download for concluído. Este banco de dados não oferece tanta flexibilidade em relação ao download seletivo em comparação com BOLD ou NCBI. Em vez disso, os dados do EMBL estão estruturados em 15 divisões fiscais, que podem ser descarregadas separadamente. A divisão tributária a ser baixada pode ser especificada usando o parâmetro --taxon . Como cada divisão fiscal é dividida em vários arquivos, é fornecido um * após o nome para baixar todos os arquivos. Os usuários também podem baixar um arquivo específico escrevendo o nome do arquivo por extenso. Uma lista de todas as 15 opções de divisão tributária é fornecida abaixo. O diretório de saída e o nome do arquivo podem ser especificados usando o parâmetro --output .
Lista de divisões fiscais:
crabs --download-embl --taxon 'mam*' --output crabs_testing/embl_mam.fasta
--download-mitofish CRABS também pode baixar o banco de dados MitoFish. Este banco de dados é um arquivo fasta único de duas linhas. O diretório de saída e o nome do arquivo podem ser especificados usando o parâmetro --output .
crabs --download-mitofish --output crabs_testing/mitofish.fasta

--download-ncbi As sequências do banco de dados NCBI são baixadas através do Entrez Programming Utilities. O NCBI permite o download de dados de vários bancos de dados, que os usuários podem especificar com o parâmetro --database . Para a maioria dos usuários, o banco de dados --database nucleotide será mais apropriado para construir um banco de dados de referência local.
Para especificar os dados a serem baixados do NCBI, os usuários realizam uma busca por meio do parâmetro --query . Elaborar boas pesquisas no NCBI pode ser difícil. Uma boa maneira de criar uma consulta de pesquisa é usar a janela de pesquisa da página da web do NCBI. A partir deste link, primeiro faça uma pesquisa inicial e pressione Enter. Isso o levará à página de resultados, onde poderá refinar ainda mais sua pesquisa. Na captura de tela abaixo, refinamos ainda mais a pesquisa, limitando o comprimento da sequência entre 100 e 25.000 pb e incorporando apenas sequências mitocondriais. Os usuários podem copiar e colar o texto na caixa "Detalhes da pesquisa" no site e fornecê-lo entre aspas no parâmetro --query . Outro benefício de usar a janela de pesquisa da página da web do NCBI é que a página da web exibirá quantas sequências correspondem à sua consulta de pesquisa, que deve corresponder ao número de sequências relatadas pelo CRABS. Esta página da web fornece mais um breve tutorial sobre como usar a função de pesquisa na página do NCBI que nossa equipe escreveu para obter informações extras.
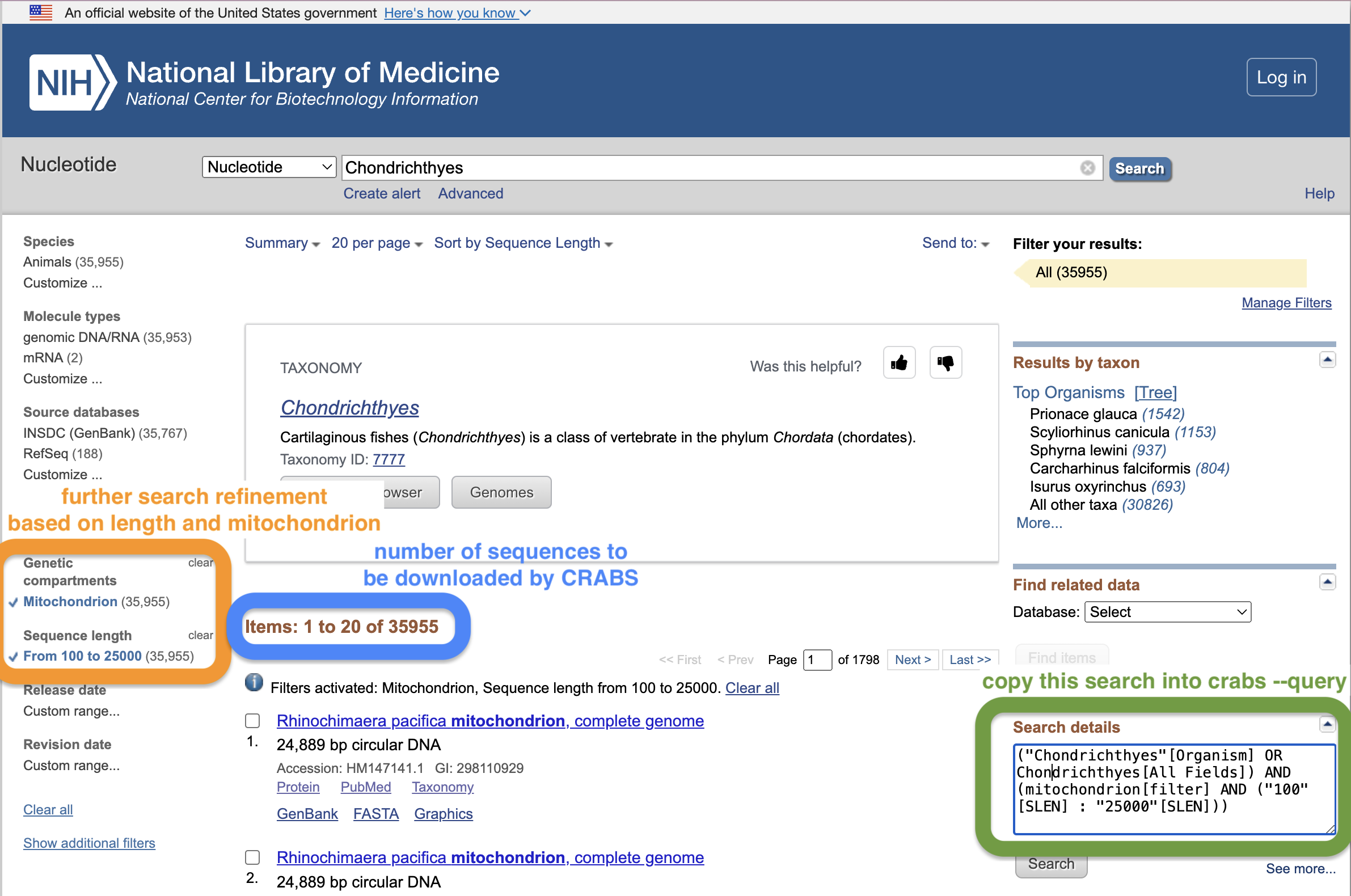
Além da consulta de pesquisa ( --query ), os usuários podem restringir ainda mais o termo de pesquisa baixando dados de sequência para uma lista de espécies usando o parâmetro --species . O parâmetro --species recebe uma sequência de entrada de nomes de espécies separados por + ou um arquivo .txt de entrada com um único nome de espécie por linha no documento. O parâmetro --batchsize oferece aos usuários a opção de baixar sequências em lotes de N do site do NCBI. Este parâmetro é padronizado como 5.000. Não é recomendado aumentar este valor acima de 5.000, pois os servidores NCBI provavelmente desconectarão o download se muitas sequências forem baixadas de uma só vez. O parâmetro --email permite que os usuários especifiquem seu endereço de e-mail, necessário para acessar os servidores NCBI. Finalmente, o diretório de saída e o nome do arquivo podem ser especificados usando o parâmetro --output .
crabs --download-ncbi --query '("Chondrichthyes"[Organism] OR Chondrichthyes[All Fields]) AND (mitochondrion[filter] AND ("100"[SLEN] : "25000"[SLEN]))' --output crabs_testing/ncbi_chondrichthyes.fasta --email [email protected] --database nucleotide

--import Depois que os dados dos repositórios online forem baixados, os arquivos precisarão ser importados para o CRABS usando a função --import . O formato CRABS constitui uma única linha delimitada por tabulação por sequência contendo todas as informações, incluindo (i) ID da sequência, (ii) nome taxonômico analisado a partir do download inicial, (iii) número de ID do táxon NCBI, (iv) linhagem taxonômica de acordo com a taxonomia NCBI e (v) a sequência. O CRABS tentará obter o número de acesso do NCBI para cada sequência como um ID de sequência. Se a sequência não contiver um número de acesso, ou seja, não for depositada no NCBI, o CRABS irá gerar IDs de sequência únicos usando o seguinte formato: crabs_*[num]*_taxonomic_name . O formato do documento de entrada é especificado usando o parâmetro --import-format e especifica o nome do repositório do qual os dados foram baixados, ou seja, BOLD , EMBL , MITOFISH ou NCBI . A linhagem taxonômica que o CRABS cria é baseada na taxonomia do NCBI e o CRABS requer os três arquivos baixados usando a função --download-taxonomy , ou seja, --names , --nodes e --acc2tax . A partir da versão v 1.0.0 , o CRABS é capaz de resolver sinônimos e nomes não aceitos para incorporar um maior número de sequências e diversidade na base de dados de referência local. As classificações taxonômicas a serem incluídas na linhagem taxonômica podem ser especificadas usando os parâmetros --ranks . Embora qualquer classificação taxonômica possa ser incluída, recomendamos usar a seguinte entrada para incluir todas as informações necessárias para a maioria dos classificadores taxonômicos --ranks 'superkingdom;phylum;class;order;family;genus;species' . O arquivo de saída pode ser especificado usando o parâmetro --output e é um arquivo .txt simples. Na janela do Terminal, o CRABS imprime os resultados do número de sequências importadas, bem como quaisquer sequências para as quais nenhuma linhagem taxonômica pôde ser gerada.
crabs --import --import-format bold --input crabs_testing/bold_Elasmobranchii.fasta --names crabs_testing/names.dmp --nodes crabs_testing/nodes.dmp --acc2tax crabs_testing/nucl_gb.accession2taxid --output crabs_testing/crabs_bold.txt --ranks 'superkingdom;phylum;class;order;family;genus;species'

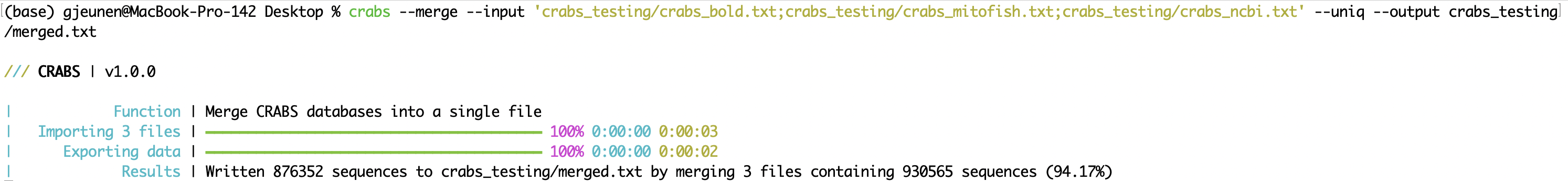
--merge Quando dados de sequência de vários repositórios online são baixados, os arquivos podem ser mesclados em um único arquivo após a importação (consulte 5.2.1 --import ) usando a função --merge . Os arquivos de entrada a serem mesclados podem ser inseridos usando o parâmetro --input , com arquivos separados por ; . É possível que uma sequência tenha sido baixada diversas vezes quando depositada em vários repositórios online. O uso do parâmetro --uniq retém apenas uma única versão de cada número de acesso. O arquivo de saída pode ser especificado usando o parâmetro --output . Na janela do Terminal, o CRABS imprime os resultados do número de sequências mescladas, bem como o número de sequências retidas ao usar o parâmetro --uniq .
crabs --merge --input 'crabs_testing/crabs_bold.txt;crabs_testing/crabs_mitofish.txt;crabs_testing/crabs_ncbi.txt' --uniq --output crabs_testing/merged.txt
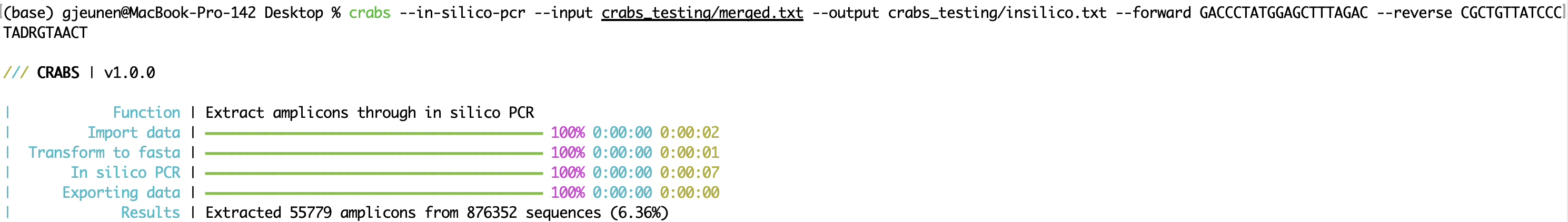
CRABS extrai a região amplificada do conjunto de primers conduzindo uma PCR in silico (função: --in-silico-pcr ). CRABS usa cutadapt v 4.4 para PCR in silico para aumentar a velocidade de execução do código python tradicional. Os nomes dos arquivos de entrada e saída podem ser especificados usando os parâmetros ' --input ' e ' --output ', respectivamente. Tanto o primer direto quanto o reverso devem ser fornecidos na direção 5'-3' usando os parâmetros ' --forward ' e ' --reverse ', respectivamente. CRABS irá complementar o primer reverso. A partir da versão v 1.0.0 , o CRABS é capaz de reter códigos de barras em ambas as direções usando uma única análise de PCR in silico . Assim, nenhuma etapa de complementação reversa e a nova execução da PCR in silico são realizadas, aumentando significativamente a velocidade de execução. Para reter sequências para as quais nenhuma região de ligação de primer foi encontrada, um arquivo de saída pode ser especificado para o parâmetro --untrimmed . O número máximo permitido de incompatibilidades encontradas nas regiões de ligação do primer pode ser especificado usando o parâmetro --mismatch , com uma configuração padrão de 4. Finalmente, a análise de PCR in silico pode ser multithread no CRABS. Por padrão, o número máximo de threads está sendo usado, mas os usuários podem especificar o número de threads a serem usados com o parâmetro --threads .
crabs --in-silico-pcr --input crabs_testing/merged.txt --output crabs_testing/insilico.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT
É prática comum remover regiões de ligação ao iniciador das sequências de referência quando depositadas numa base de dados online. Portanto, quando a sequência de referência foi gerada usando o mesmo iniciador direto e/ou reverso pesquisado na função --in-silico-pcr , a função --in-silico-pcr não terá conseguido recuperar a região amplicon do sequência de referência. Para levar em conta esta possibilidade, o CRABS tem a opção de executar um Alinhamento Global Pairwise, implementado usando VSEARCH v 2.16.0 , para extrair regiões de amplificação para as quais a sequência de referência não contém as regiões completas de ligação direta e reversa ao primer. Para fazer isso, a função --pairwise-global-alignment pega o arquivo de banco de dados baixado originalmente usando o parâmetro --input . O banco de dados a ser pesquisado é o arquivo de saída do --in-silico-pcr e pode ser especificado usando o parâmetro --amplicons . O arquivo de saída pode ser especificado usando o parâmetro --output . As sequências de primers, usadas apenas para calcular o comprimento do par de bases, podem ser definidas com os parâmetros --forward e --reverse . Como a função --pairwise-global-alignment pode levar muito tempo para ser executada em bancos de dados grandes, o comprimento da sequência pode ser restrito para acelerar o processo usando o parâmetro --size-select . A porcentagem mínima de identidade e cobertura de consulta pode ser especificada usando os parâmetros --percent-identity e --coverage , respectivamente. --percent-identity deve ser fornecido como um valor percentual entre 0 e 1 (por exemplo, 95% = 0,95), enquanto --coverage deve ser fornecido como um valor percentual entre 0 e 100 (por exemplo, 95% = 95). Por padrão, a função --pairwise-global-alignment é restrita para reter sequências onde as sequências de primer não estão totalmente presentes na sequência de referência (alinhamento começando ou terminando dentro do comprimento do primer direto ou reverso). Quando o parâmetro --all-start-positions é fornecido, acertos positivos serão incluídos quando o alinhamento for encontrado fora do intervalo das regiões de ligação do primer (perdido pela função --in-silico-pcr devido a muitas incompatibilidades no região de ligação ao primer). Não recomendamos o uso de --all-start-positions , pois é muito improvável que um código de barras seja amplificado usando o conjunto de primers especificado da função --in-silico-pcr quando mais de 4 incompatibilidades estiverem presentes no primer- regiões de ligação.
crabs --pairwise-global-alignment --input crabs_testing/merged.txt --amplicons crabs_testing/insilico.txt --output crabs_testing/aligned.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT --size-select 10000 --percent-identity 0.95 --coverage 0.95
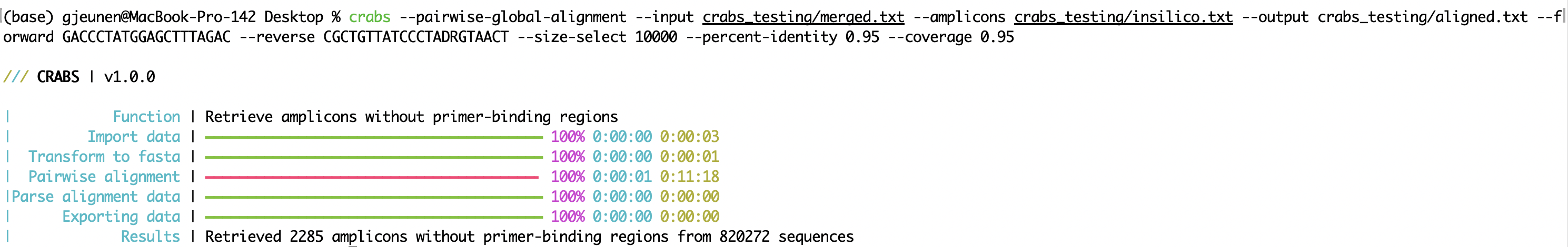
--pairwise-global-alignment A função --pairwise-global-alignment pode levar uma quantidade substancial de tempo para ser executada quando o CRABS está processando arquivos de sequência grandes, mesmo que multithreading seja suportado. Desde a atualização para CRABS v 1.0.0 , uma estrutura de arquivo idêntica existe de --import para --export , permitindo assim que funções sejam executadas em qualquer ordem. Embora ainda recomendemos seguir a ordem do fluxo de trabalho CRABS, a função --pairwise-global-alignment pode ser significativamente acelerada ao executar as funções --dereplicate e --filter antes da função --in-silico-pcr . Ao executar essas etapas de curadoria antes de --in-silico-pcr , o número de sequências necessárias a serem processadas pelo CRABS para a função --pairwise-global-alignment será significativamente reduzido.
NOTA 1 : ao executar a função --filter antes de --in-silico-pcr , certifique-se de omitir quaisquer parâmetros que estejam impactando diretamente a sequência, pois --filter baseará isso em toda a sequência e não no amplicon extraído . Portanto, omita os seguintes parâmetros: --minimum-length , --maximum-length , --maximum-n .
NOTA 2 : ao executar as funções --dereplicate e --filter antes de --in-silico-pcr , seria aconselhável executar ambas as funções novamente após --pairwise-global-alignment , pois o banco de dados poderia ser mais curado agora que os amplicons sejam extraídos.
Uma vez que todos os códigos de barras potenciais para o conjunto de primers tenham sido extraídos pelas funções --in-silico-pcr e --pairwise-global-alignment , o banco de dados de referência local pode passar por curadoria e subconjunto adicionais dentro do CRABS usando várias funções, incluindo --dereplicate , --filter e --subset .
--dereplicate O primeiro método de curadoria é desreplicar o banco de dados de referência local usando a função --dereplicate . É possível que, para certos táxons, vários códigos de barras idênticos estejam contidos no banco de dados de referência local neste momento. Isso pode ocorrer quando diferentes grupos de pesquisa depositaram sequências idênticas ou se a variação intraespecífica entre sequências de um táxon não estiver contida no código de barras extraído. É melhor remover esses códigos de barras de referência idênticos para acelerar a atribuição de taxonomia, bem como melhorar os resultados da atribuição de taxonomia (especialmente para classificadores taxonômicos que fornecem um número limitado de resultados, ou seja, BLAST).
Os arquivos de entrada e saída podem ser especificados usando os parâmetros --input e --output , respectivamente. CRABS oferece três métodos de desreplicação, que podem ser especificados usando o parâmetro --dereplication-method , incluindo:
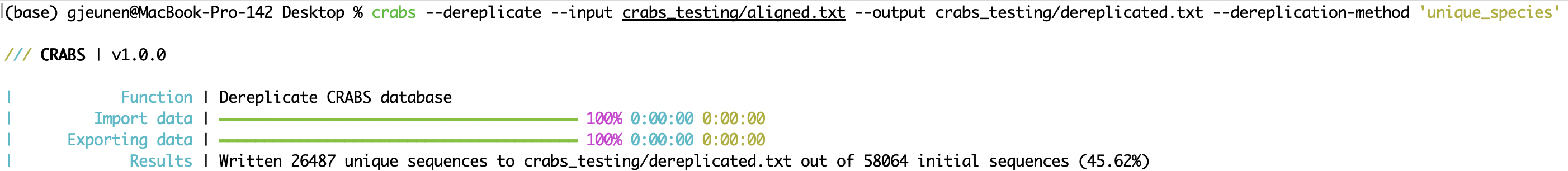
crabs --dereplicate --input crabs_testing/aligned.txt --output crabs_testing/dereplicated.txt --dereplication-method 'unique_species'
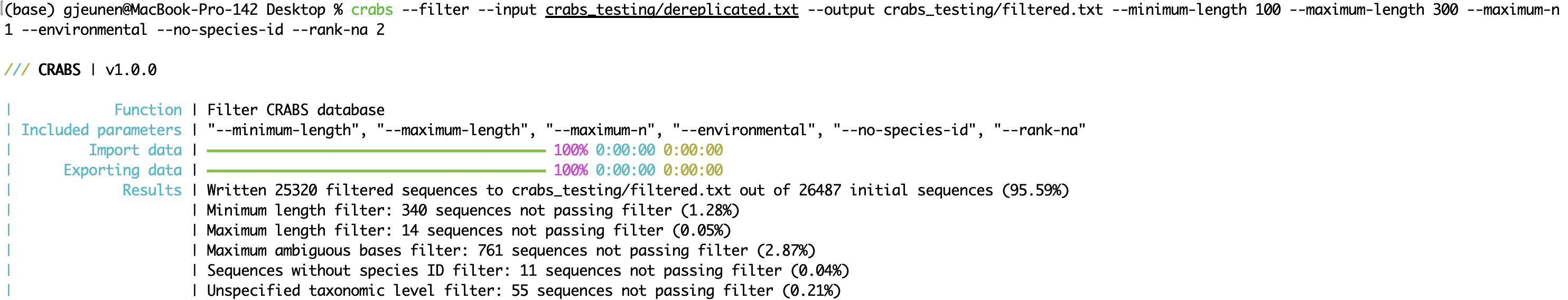
--filter O segundo método de curadoria é filtrar o banco de dados de referência local usando vários parâmetros usando a função --filter . Os arquivos de entrada e saída podem ser especificados usando os parâmetros --input e --output , respectivamente. Da versão v 1.0.0 . CRABS incorpora a filtragem baseada em seis parâmetros, incluindo:
--minimum-length : comprimento mínimo de sequência para um amplicon ser retido no banco de dados;--maximum-length : comprimento máximo da sequência para um amplicon ser retido no banco de dados;--maximum-n : descarta amplicons com N ou mais bases ambíguas ( N );--environmental : descarta sequências ambientais do banco de dados;--no-species-id : descarta sequências para as quais nenhum nome de espécie está disponível;--rank-na : descarta sequências com N ou mais níveis taxonômicos não especificados. crabs --filter --input crabs_testing/dereplicated.txt --output crabs_testing/filtered.txt --minimum-length 100 --maximum-length 300 --maximum-n 1 --environmental --no-species-id --rank-na 2
--subset O terceiro e último método de curadoria incorporado no CRABS é subdividir o banco de dados de referência local para incluir (parâmetro: --include ) ou excluir (parâmetro: --exclude ) táxons específicos usando a função --subset . Esta função permite a remoção de códigos de barras de referência de grupos taxonômicos que não interessam à questão de pesquisa. Estes grupos taxonômicos poderiam ter sido incorporados ao banco de dados de referência local devido à potencial amplificação inespecífica do conjunto de primers. Outro caso de uso para --subset é remover sequências erradas conhecidas.
Para classificadores taxonômicos baseados em aprendizado de máquina (IDTAXA) ou distância k-mer (SINTAX), pode ser benéfico subdividir o banco de dados de referência incluindo apenas táxons conhecidos por ocorrerem na região onde as amostras foram coletadas e excluir espécies estreitamente relacionadas conhecidas não ocorrer na região para aumentar a resolução taxonômica obtida desses classificadores e obter melhores resultados de atribuição de taxonomia.
Os arquivos de entrada e saída podem ser especificados usando os parâmetros --input e --output , respectivamente. Os parâmetros --include e --exclude podem conter uma lista de táxons separados por ; ou um arquivo .txt contendo um único nome de táxon por linha.
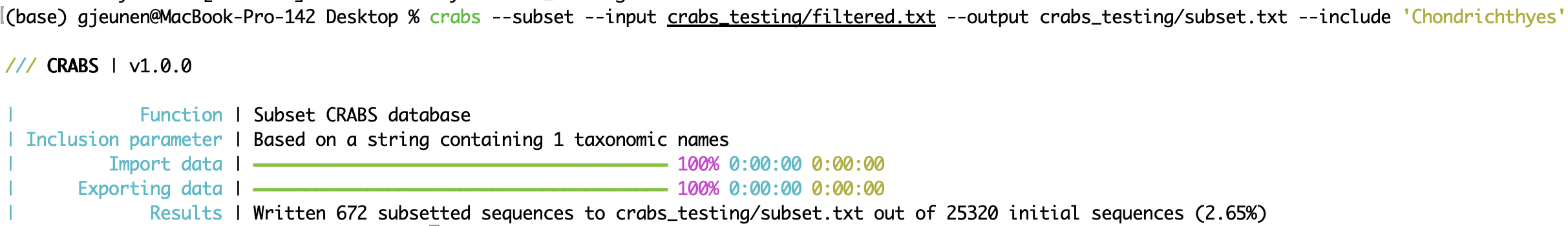
crabs --subset --input crabs_testing/filtered.txt --output crabs_testing/subset.txt --include 'Chondrichthyes'
Uma vez finalizado o banco de dados de referência, ele pode ser exportado em vários formatos para acomodar as especificações exigidas pela maioria das ferramentas de software que atribuem taxonomia aos dados metagenômicos. Os arquivos de entrada e saída podem ser especificados usando os parâmetros --input e --output , respectivamente. A partir da versão v 1.0.0 , o CRABS incorpora a formatação do banco de dados de referência para seis classificadores diferentes (parâmetro: --export-format ), incluindo:
--export-format 'sintax' : O classificador SINTAX é incorporado em USEARCH e VSEARCH;--export-format 'rdp' : O classificador RDP é um programa independente amplamente utilizado em estudos de microbiomas;--export-format 'qiime-fasta' e --export-format 'qiime-text' : Pode ser usado para atribuir um ID taxonômico em QIIME e QIIME2;--export-format 'dada2-species' e --export-format 'dada2-taxonomy' : Pode ser usado para atribuir um ID taxonômico em DADA2;--export-format 'idt-fasta' e --export-format 'idt-text' : O classificador IDTAXA é um algoritmo de aprendizado de máquina incorporado no pacote DECIPHER R;--export-format 'blast-notax' : cria um banco de dados de referência de explosão local para blastn e megablast, onde a saída não fornece um ID taxonômico, mas lista o número de acesso;--export-format 'blast-tax' : cria um banco de dados de referência de explosão local para BLASTN e Megablast, onde a saída fornece o ID taxonômico e o número de acesso. crabs --export --input crabs_testing/subset.txt --output crabs_testing/BLAST_TAX_CHONDRICHTHYES --export-format 'blast-tax'

Ao exportar o banco de dados de referência local para um único formato (exceto os classificadores em que o banco de dados de referência é dividido em vários arquivos, ou seja, qiime, dada2, idtaxa) será suficiente para a maioria dos usuários, um loop simples para exportar o local para exportar o local Banco de dados de referência a vários formatos se os usuários desejarem comparar resultados entre diferentes classificadores taxonômicos. Um exemplo é fornecido abaixo para exportar o banco de dados de referência local nos formatos Sintax, RDP e IDTAXA.
for format in sintax.fasta rdp.fasta idt-fasta.fasta idt-text.txt; do crabs --export --input crabs_testing/subset.txt --output crabs_testing/chondrichthyes_${format} --export-format ${format%%.*}; done

Depois que o banco de dados de referência é finalizado, os caranguejos podem executar cinco funções de pós-processamento para explorar e fornecer uma visão geral resumida do banco de dados de referência local, incluindo (i) --diversity-figure , (ii) --amplicon-length-figure , ( iii) --phylogenetic-tree , (iv) --amplification-efficiency-figure e (v) --completeness-table .
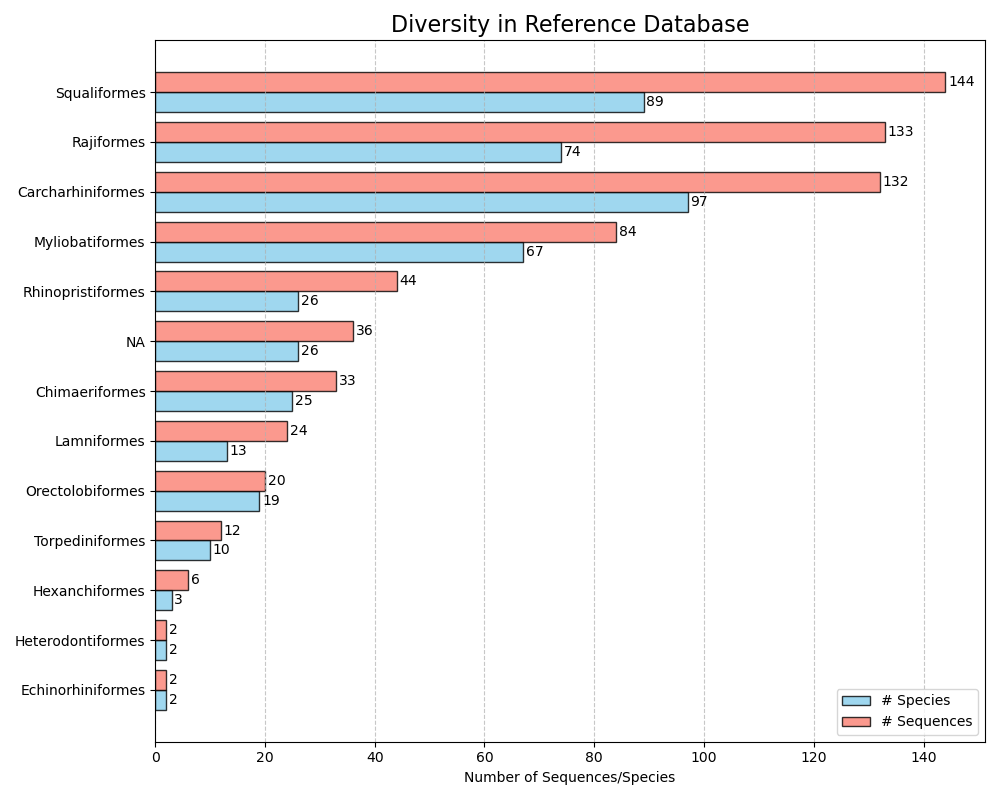
--diversity-figure A função --diversity-figure produz um gráfico de barra horizontal com número de espécies (em azul) e número de sequências (em laranja) por cada grupo taxonômico no banco de dados de referência. O usuário pode especificar a classificação taxonômica para dividir o banco de dados de referência com o parâmetro --tax-level . O nível tributário é o número da classificação em que apareceu durante a função --import . Por exemplo, se --ranks 'superkingdom;phylum;class;order;family;genus;species' foi utilizado durante a divisão --import com base na superdomínio exigiria --tax-level 1 , Phylum = --tax-level 2 , Class = --tax-level 3 , etc. O arquivo de entrada no formato dos caranguejos pode ser especificado usando o parâmetro --input . A figura, no formato .png, será gravada no arquivo de saída, que pode ser especificado usando o parâmetro --output .
crabs --diversity-figure --input crabs_testing/subset.txt --output crabs_testing/diversity-figure.png --tax-level 4
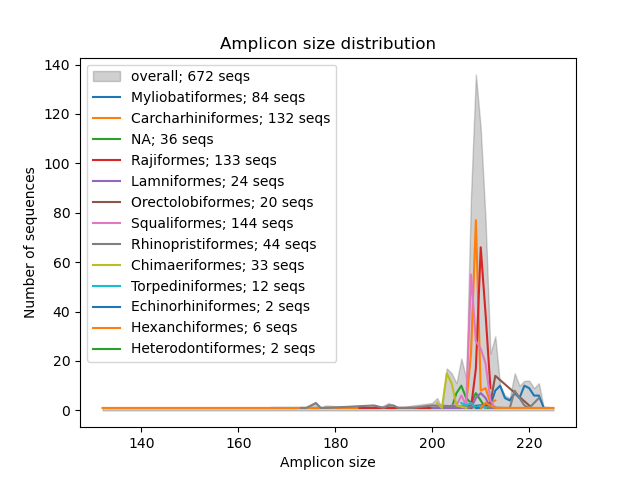
--amplicon-length-figure A função --amplicon-length-figure produz um gráfico de linha exibindo o intervalo do comprimento do amplicon. O intervalo geral no comprimento do amplicon em todas as seqüências no banco de dados de referência é exibido em uma cor cinza sombreada, enquanto os resultados divididos por grupo taxonômico (parâmetro: --tax-level ) são sobrepostos por linhas coloridas. Além disso, a legenda exibe o número de seqüências atribuídas a cada um dos grupos taxonômicos e o número total de sequências no banco de dados de referência. O arquivo de entrada no formato dos caranguejos pode ser especificado usando o parâmetro --input . A figura, no formato .png, será gravada no arquivo de saída, que pode ser especificado usando o parâmetro --output .
crabs --amplicon-length-figure --input crabs_testing/subset.txt --output crabs_testing/amplicon-length-figure.png --tax-level 4
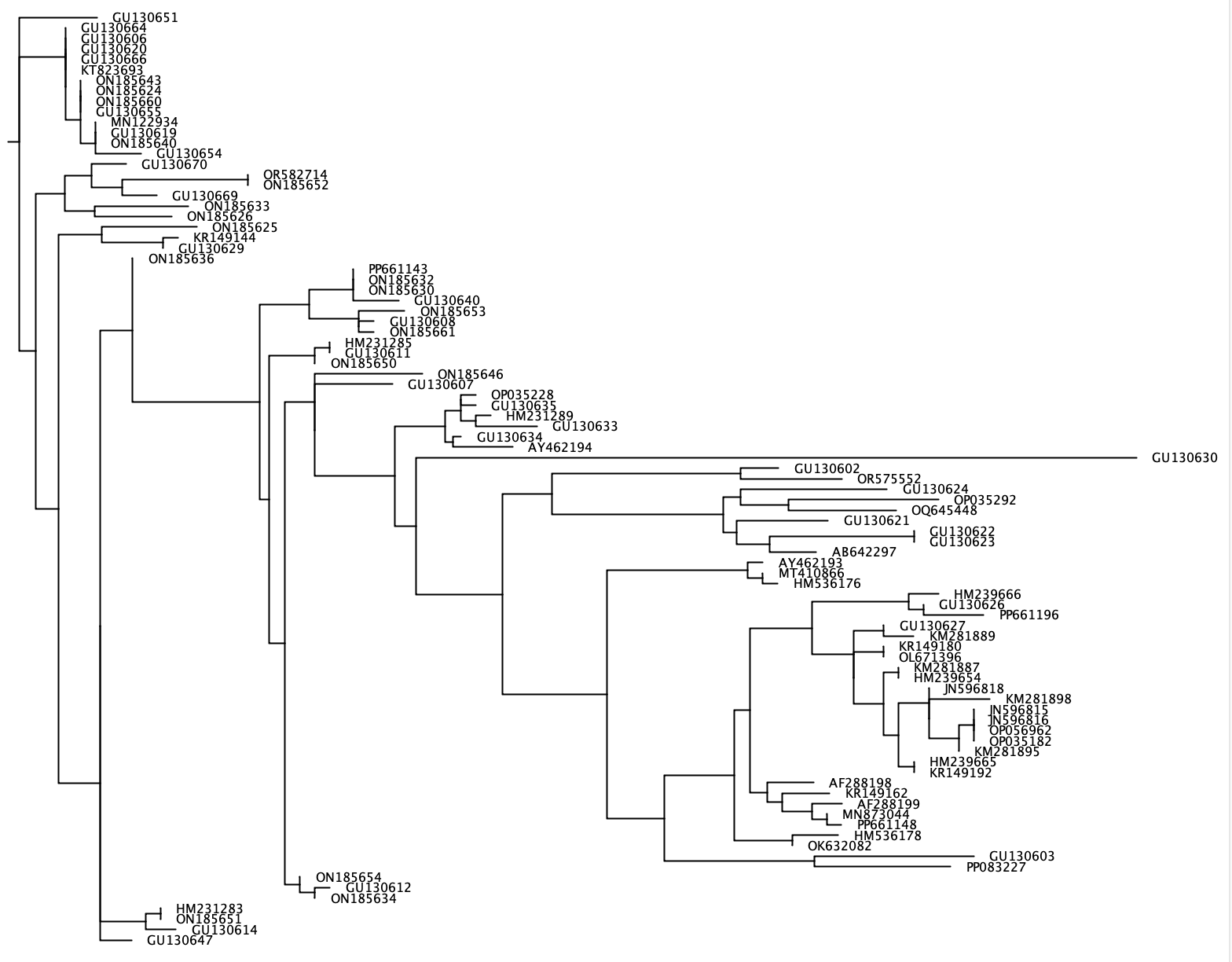
--phylogenetic-tree A função --phylogenetic-tree gerará uma árvore filogenética para uma lista de espécies de interesse. Esta lista de espécies de interesse pode ser importada usando o parâmetro --species e consiste em uma sequência de entrada separada por + ou um arquivo .txt com um nome de espécie única em cada linha. Para cada espécie de interesse, as seqüências serão extraídas do banco de dados de referência que compartilham uma classificação taxonômica definida pelo usuário (parâmetro: --tax-level ) com a espécie de interesse. Os caranguejos gerarão um alinhamento de todas as seqüências extraídas usando o CLUSTALW2 V 2.1 e gerarão uma árvore filogenética de união de vizinhos usando o FastTree. Esta árvore filogenética no formato Newick será gravada no arquivo de saída usando o parâmetro --output e pode ser visualizada em programas de software como Figtree ou Geneious. Como uma árvore filogenética separada será gerada para cada espécie de interesse, --output recebe um nome de arquivo genérico, enquanto o arquivo de saída exato conterá esse nome genérico seguido por '_species_name.tree'.
crabs --phylogenetic-tree --input crabs_testing/subset.txt --output crabs_testing/phylo --tax-level 4 --species 'Carcharodon carcharias+Squalus acanthias'
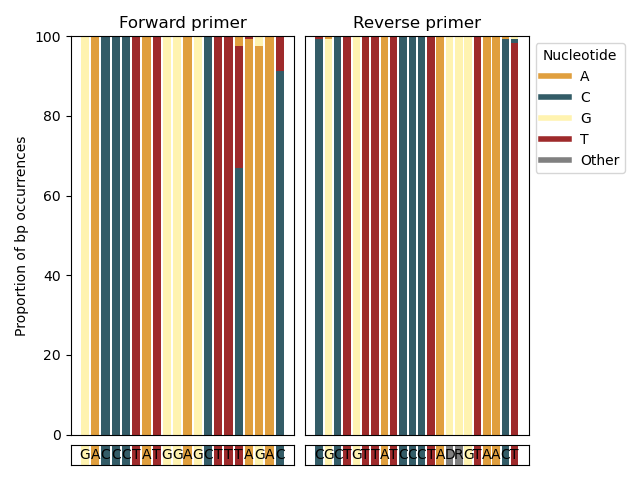
--amplification-efficiency-figure A função --amplification-efficiency-figure produzirá um gráfico de barras, exibindo a proporção de ocorrência de pares de bases nas regiões de ligação a iniciadores para um grupo taxonômico especificado pelo usuário, visualizando os lugares nas regiões de ligação a iniciadores avançadas e reversas onde as incompatibilidades Pode estar ocorrendo no grupo taxonômico de juros, potencialmente influenciando a eficiência da amplificação. A função --amplification-efficiency-figure FUNCIONATE UM BATO DE REFERÊNCIA FINAL FORMATIDO DE CARCARS como entrada usando o parâmetro --amplicons . Para encontrar as informações sobre as regiões de ligação ao iniciador para cada sequência no arquivo de entrada, as seqüências inicialmente baixadas após a importação precisam ser fornecidas usando o parâmetro --input . As sequências de iniciadores avançadas e reversas (na direção 5 ' -3') são fornecidas usando os parâmetros --forward e --reverse . O nome do grupo taxonômico de juros pode ser fornecido usando o parâmetro --tax-group e pode ser definido em qualquer nível taxonômico incorporado no arquivo de entrada. Finalmente, a figura no formato .png será gravada no arquivo de saída especificado pelo parâmetro --output .
crabs --amplification-efficiency-figure --input crabs_testing/merged.txt --amplicons crabs_testing/subset.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT --output crabs_testing/amplification-efficiency.png --tax-group Carcharhiniformes
--completeness-table A função --completeness-table produzirá uma tabela delimitada por TAB (Parâmetro: --output ) com informações sobre uma lista de espécies de interesse. Esta lista de espécies de interesse pode ser importada usando o parâmetro --species e consiste em uma sequência de entrada separada por + ou um arquivo .txt com um nome de espécie única em cada linha. Uma linhagem taxonômica será gerada para cada espécie de interesse usando os arquivos ' nomes.dmp ' e ' nós.dmp ' baixados usando a função --download-taxonomy usando os parâmetros --names e --nodes , respectivamente. A tabela de saída terá 10 colunas, fornecendo as seguintes informações:
crabs --completeness-table --input crabs_testing/subset.txt --output crabs_testing/completeness.txt --names crabs_testing/names.dmp --nodes crabs_testing/nodes.dmp --species 'Carcharodon carcharias+Squalus acanthias'
crabs --version v 1.0.6 : Corrigir de bug -> A análise melhorada de cabeçalhos em negrito durante --import .crabs --version v 1.0.5 : Corrigir de bug -> Adicionado uma restrição de comprimento ao ID SEQ ao criar bancos de dados de explosão, conforme necessário para o software BLAST+.crabs --version v 1.0.4 : Informações adicionadas-> Forneceu informações corretas sobre a entrada de valor para --pairwise-global-alignment --coverage --percent-identity .crabs --version v 1.0.3 : Corrigação por bug -> Verificando a resposta do servidor NCBI 3 vezes antes de abortar a análise.crabs --version v 1.0.2 : Corrigir de bug -> Capaz de relatar quando 0 sequências são retornadas após a análise.crabs --version v 1.0.1 : Correção de bug -> Consulta de construção bem -sucedida do NCBI usando o parâmetro --species .


