nucleotide transformer
1.0.0
Bem-vindo a este repositório InstaDeep Github, onde estão apresentados:
Estamos entusiasmados em abrir o código-fonte desses trabalhos e fornecer à comunidade acesso ao código e aos pesos pré-treinados para esses nove modelos de linguagem genômica e dois modelos de segmentação. Os modelos do projeto nucleotide transformer foram desenvolvidos em colaboração com Nvidia e TUM, e os modelos foram treinados em nós DGX A100 em Cambridge-1. O modelo do projeto Agro nucleotide transformer foi desenvolvido em colaboração com o Google, e o modelo treinado em aceleradores TPU-v4.
No geral, nossos trabalhos fornecem novos insights relacionados ao pré-treinamento e aplicação de modelos fundamentais de linguagem, bem como ao treinamento de modelos que os utilizam como codificador de backbone, para genômica com amplas oportunidades de suas aplicações no campo.
Neste repositório, você encontrará o seguinte:
Em comparação com outras abordagens, nossos modelos não apenas integram informações de genomas de referência únicos, mas também aproveitam sequências de DNA de mais de 3.200 genomas humanos diversos, bem como 850 genomas de uma ampla gama de espécies, incluindo organismos modelo e não modelo. Através de uma avaliação robusta e extensa, mostramos que estes grandes modelos fornecem uma previsão do fenótipo molecular extremamente precisa em comparação com os métodos existentes.
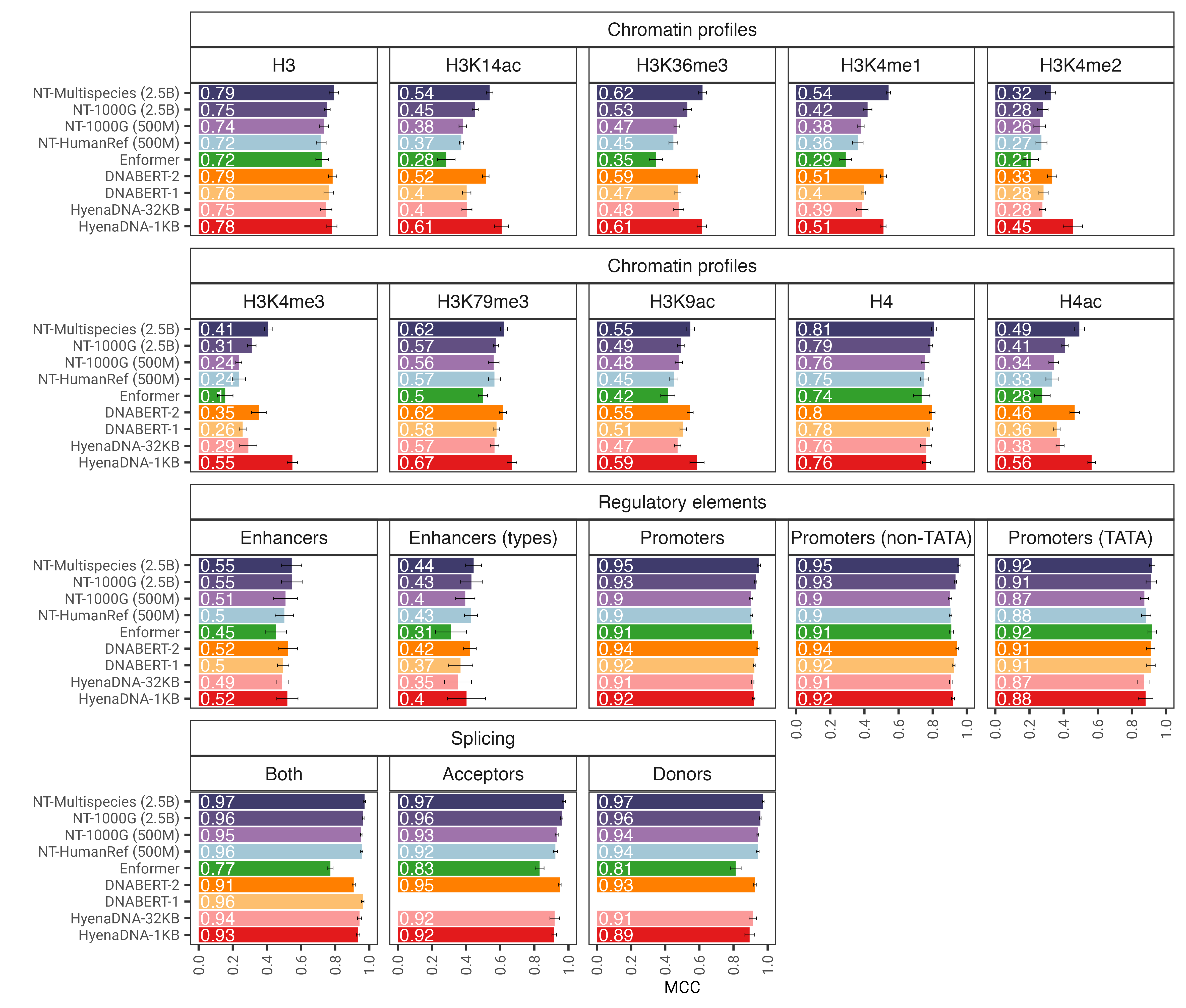
Figura 1: O modelo nucleotide transformer prevê com precisão diversas tarefas genômicas após o ajuste fino. Mostramos os resultados de desempenho em tarefas posteriores para modelos de transformadores ajustados. As barras de erro representam 2 SDs derivados da validação cruzada de 10 vezes.
Neste trabalho apresentamos um novo modelo fundamental de linguagem grande treinado em genomas de referência de 48 espécies de plantas com foco predominante em espécies agrícolas. Avaliamos o desempenho do AgroNT em diversas tarefas de previsão, desde características regulatórias, processamento de RNA e expressão gênica, e mostramos que o AgroNT pode obter desempenho de última geração.
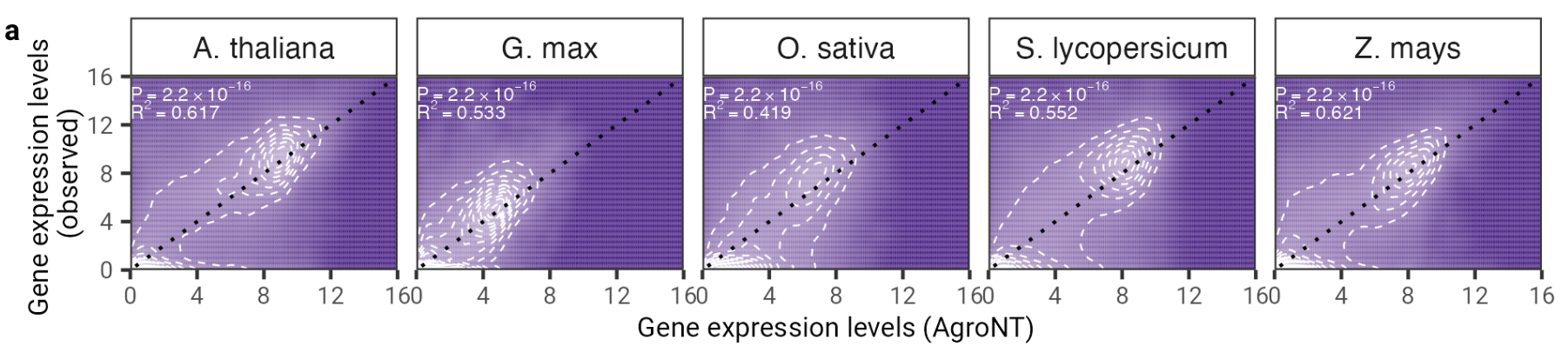
Figura 2: AgroNT fornece previsão de expressão gênica em diferentes espécies de plantas. A previsão da expressão gênica em genes de resistência em todos os tecidos está correlacionada com os níveis de expressão gênica observados. O coeficiente de determinação (R 2 ) de um modelo linear e os valores P associados entre os valores previstos e observados são mostrados.
Para usar o código e os modelos pré-treinados, basta:
pip install . .Você pode então baixar e fazer a inferência com qualquer um dos nossos nove modelos em apenas algumas linhas de códigos:
import haiku as hk
import jax
import jax . numpy as jnp
from nucleotide_transformer . pretrained import get_pretrained_model
# Get pretrained model
parameters , forward_fn , tokenizer , config = get_pretrained_model (
model_name = "500M_human_ref" ,
embeddings_layers_to_save = ( 20 ,),
max_positions = 32 ,
)
forward_fn = hk . transform ( forward_fn )
# Get data and tokenize it
sequences = [ "ATTCCGATTCCGATTCCG" , "ATTTCTCTCTCTCTCTGAGATCGATCGATCGAT" ]
tokens_ids = [ b [ 1 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens_str = [ b [ 0 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens = jnp . asarray ( tokens_ids , dtype = jnp . int32 )
# Initialize random key
random_key = jax . random . PRNGKey ( 0 )
# Infer
outs = forward_fn . apply ( parameters , random_key , tokens )
# Get embeddings at layer 20
print ( outs [ "embeddings_20" ]. shape )Os nomes dos modelos suportados são:
Você também pode executar nossos modelos e encontrar mais exemplos de código no Google Colab
O código roda tanto em GPU quanto em TPU graças ao Jax!
Nossos modelos nucleotide transformer v2 da segunda versão incluem uma série de mudanças arquitetônicas que se mostraram mais eficientes: em vez de usar embeddings posicionais aprendidos, usamos Rotary Embeddings que são usados em cada camada de atenção e unidades lineares fechadas com ativações swish sem viés. Esses modelos aprimorados também aceitam sequências de até 2.048 tokens, levando a uma janela de contexto mais longa de 12kbp. Inspirados pelas leis de escalonamento da Chinchilla, também treinamos nossos modelos NT-v2 em nosso conjunto de dados multiespécies para maior duração (tokens de 300B para os modelos 50M e 100M; tokens de 1T para o modelo 250M e 500M) em comparação com os modelos v1 (tokens de 300B para todos os quatro modelos).
As camadas do transformador são indexadas em 1, o que significa que chamar get_pretrained_model com os argumentos model_name="500M_human_ref" e embeddings_layers_to_save=(1, 20,) resultará na extração de embeddings após a primeira e a 20ª camada do transformador. Para transformadores que utilizam o cabeçote Roberta LM, é prática comum extrair os embeddings finais após a norma da primeira camada do cabeçote LM, e não após o último bloco do transformador. Portanto, se get_pretrained_model for chamado com os seguintes argumentos embeddings_layers_to_save=(24,) , os embeddings não serão extraídos após a camada final do transformador, mas sim após a norma da primeira camada do cabeçote LM.
Os modelos SegmentNT aproveitam um nucleotide transformer (NT) do qual removemos o cabeçote do modelo de linguagem e o substituímos por um cabeçote de segmentação U-Net unidimensional para prever a localização de vários tipos de elementos genômicos em uma sequência com uma resolução de nucleotídeo único. Apresentamos duas variantes de modelo diferentes em 14 classes diferentes de elementos genômicos humanos em sequências de entrada de até 30kb. Estes incluem genes (genes codificadores de proteínas, lncRNAs, 5'UTR, 3'UTR, exon, íntron, receptores de splice e locais doadores) e reguladores (sinal poliA, promotores e intensificadores invariantes de tecido e específicos de tecido, e CTCF- ligado sites) elementos. SegmentNT alcança desempenho superior em relação à arquitetura de segmentação U-Net de última geração, beneficiando-se dos pesos pré-treinados do NT e demonstra generalização zero-shot de até 50kbp.
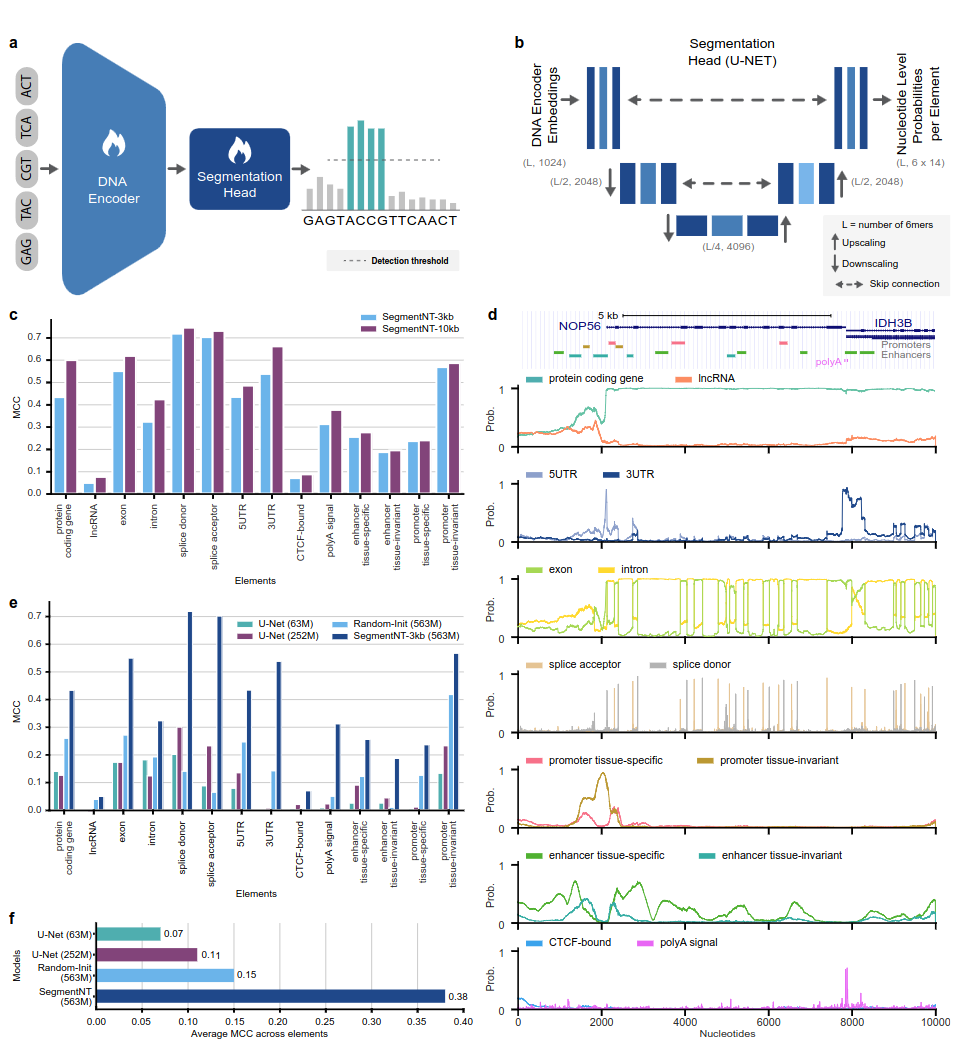
Figura 1: SegmentNT localiza elementos genômicos na resolução de nucleotídeos.
Para usar o código e os modelos pré-treinados, basta:
pip install . .Você pode então baixar e inferir uma sequência com qualquer um de nossos modelos em apenas algumas linhas de códigos:
rescaling factor é definido como aquele usado durante o treinamento. Caso você precise inferir sequências entre 30kbp e 50kbp, certifique-se de passar o argumento rescaling_factor na função get_pretrained_segment_nt_model com o valor rescaling_factor = max_num_nucleotides / max_num_tokens_nt onde num_dna_tokens_inference é o número de tokens na inferência (ou seja, 6669 para uma sequência de 40008 base pares) e max_num_tokens_nt é o número máximo de tokens nos quais o transformador de nucleotídeo backbone foi treinado, ou seja, 2048 .
? O caderno examples/inference_segment_nt.ipynb mostra como inferir em uma sequência de 50kb e traçar as probabilidades para reproduzir a Figura 3 do artigo.
? Os modelos SegmentNT não lidam com nenhum "N" na sequência de entrada porque cada nucleotídeo precisa ser tokenizado como 6-mers, o que não pode ser o caso ao usar sequências contendo um ou vários pares de bases "N".
import haiku as hk
import jax
import jax . numpy as jnp
from nucleotide_transformer . pretrained import get_pretrained_segment_nt_model
# Initialize CPU as default JAX device. This makes the code robust to memory leakage on
# the devices.
jax . config . update ( "jax_platform_name" , "cpu" )
backend = "cpu"
devices = jax . devices ( backend )
num_devices = len ( devices )
print ( f"Devices found: { devices } " )
# The number of DNA tokens (excluding the CLS token prepended) needs to be dividible by
# 2 to the power of the number of downsampling block, i.e 4.
max_num_nucleotides = 8
assert max_num_nucleotides % 4 == 0 , (
"The number of DNA tokens (excluding the CLS token prepended) needs to be dividible by"
"2 to the power of the number of downsampling block, i.e 4." )
parameters , forward_fn , tokenizer , config = get_pretrained_segment_nt_model (
model_name = "segment_nt" ,
embeddings_layers_to_save = ( 29 ,),
attention_maps_to_save = (( 1 , 4 ), ( 7 , 10 )),
max_positions = max_num_nucleotides + 1 ,
)
forward_fn = hk . transform ( forward_fn )
apply_fn = jax . pmap ( forward_fn . apply , devices = devices , donate_argnums = ( 0 ,))
# Get data and tokenize it
sequences = [ "ATTCCGATTCCGATTCCAACGGATTATTCCGATTAACCGATTCCAATT" , "ATTTCTCTCTCTCTCTGAGATCGATGATTTCTCTCTCATCGAACTATG" ]
tokens_ids = [ b [ 1 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens_str = [ b [ 0 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens = jnp . asarray ( tokens_ids , dtype = jnp . int32 )
random_key = jax . random . PRNGKey ( seed = 0 )
keys = jax . device_put_replicated ( random_key , devices = devices )
parameters = jax . device_put_replicated ( parameters , devices = devices )
tokens = jax . device_put_replicated ( tokens , devices = devices )
# Infer on the sequence
outs = apply_fn ( parameters , keys , tokens )
# Obtain the logits over the genomic features
logits = outs [ "logits" ]
# Transform them in probabilities
probabilities = jnp . asarray ( jax . nn . softmax ( logits , axis = - 1 ))[..., - 1 ]
print ( f"Probabilities shape: { probabilities . shape } " )
print ( f"Features inferred: { config . features } " )
# Get probabilities associated with intron
idx_intron = config . features . index ( "intron" )
probabilities_intron = probabilities [..., idx_intron ]
print ( f"Intron probabilities shape: { probabilities_intron . shape } " )Os nomes dos modelos suportados são:
O código roda tanto em GPU quanto em TPU graças ao Jax!
Os modelos são treinados em sequências de até 1.000 tokens, incluindo o token <CLS> anexado automaticamente ao início da sequência. O tokenizer inicia a tokenização da esquerda para a direita agrupando as letras “A”, “C”, “G” e “T” em 6-mers. A letra "N" é escolhida para não ser agrupada dentro dos k-mers, portanto sempre que o tokenizer encontrar um "N", ou se o número de nucleotídeos na sequência não for múltiplo de 6, ele irá tokenizar os nucleotídeos sem agrupar eles. Exemplos são dados abaixo:
dna_sequence_1 = "ACGTGTACGTGCACGGACGACTAGTCAGCA"
tokenized_dna_sequence_1 = [ < CLS > , < ACGTGT > , < ACGTGC > , < ACGGAC > , < GACTAG > , < TCAGCA > ]
dna_sequence_2 = "ACGTGTACNTGCACGGANCGACTAGTCTGA"
tokenized_dna_sequence_2 = [ < CLS > , < ACGTGT > , < A > , < C > , < N > , < TGCACG > , < G > , < A > , < N > , < CGACTA > , < GTCTGA > ]Todos os transformadores v1 e v2 podem, portanto, receber sequências de até 5.994 e 12.282 nucleotídeos, respectivamente, se não houver "N" em seu interior.
A coleção de modelos apresentados neste repositório está disponível nos espaços huggingface do Instadeep aqui: O espaço do nucleotide transformer e o espaço nucleotide transformer Agro!
Agradecemos a Maša Roller, bem como aos membros do Rostlab, particularmente Tobias Olenyi, Ivan Koludarov e Burkhard Rost pelas discussões construtivas que ajudaram a identificar direções interessantes de pesquisa. Além disso, estendemos a nossa gratidão a todos aqueles que depositam dados experimentais em bases de dados públicas, àqueles que mantêm essas bases de dados e àqueles que disponibilizam gratuitamente métodos analíticos e preditivos. Agradecemos também à equipe de desenvolvimento do Jax.
Se você achar este repositório útil em seu trabalho, adicione uma citação relevante a qualquer um de nossos artigos associados:
O papel nucleotide transformer :
@article { dalla2023nucleotide ,
title = { The nucleotide transformer : Building and Evaluating Robust Foundation Models for Human Genomics } ,
author = { Dalla-Torre, Hugo and Gonzalez, Liam and Mendoza Revilla, Javier and Lopez Carranza, Nicolas and Henryk Grywaczewski, Adam and Oteri, Francesco and Dallago, Christian and Trop, Evan and Sirelkhatim, Hassan and Richard, Guillaume and others } ,
journal = { bioRxiv } ,
pages = { 2023--01 } ,
year = { 2023 } ,
publisher = { Cold Spring Harbor Laboratory }
}Papel nucleotide transformer agronucleotídeo:
@article { mendoza2024foundational ,
title = { A foundational large language model for edible plant genomes } ,
author = { Mendoza-Revilla, Javier and Trop, Evan and Gonzalez, Liam and Roller, Ma{v{s}}a and Dalla-Torre, Hugo and de Almeida, Bernardo P and Richard, Guillaume and Caton, Jonathan and Lopez Carranza, Nicolas and Skwark, Marcin and others } ,
journal = { Communications Biology } ,
volume = { 7 } ,
number = { 1 } ,
pages = { 835 } ,
year = { 2024 } ,
publisher = { Nature Publishing Group UK London }
}Papel SegmentNT
@article { de2024segmentnt ,
title = { SegmentNT: annotating the genome at single-nucleotide resolution with DNA foundation models } ,
author = { de Almeida, Bernardo P and Dalla-Torre, Hugo and Richard, Guillaume and Blum, Christopher and Hexemer, Lorenz and Gelard, Maxence and Pandey, Priyanka and Laurent, Stefan and Laterre, Alexandre and Lang, Maren and others } ,
journal = { bioRxiv } ,
pages = { 2024--03 } ,
year = { 2024 } ,
publisher = { Cold Spring Harbor Laboratory }
}Se você tiver alguma dúvida ou feedback sobre o código e os modelos, sinta-se à vontade para entrar em contato conosco.
Obrigado pelo seu interesse em nosso trabalho!


