poseidon
1.0.3

Observe que o código de Poseidon foi transferido para o NextFlow, para que ainda haja alguns bugs. Sinta -se à vontade para relatar problemas!
Aqui, apresentamos Poseidon, um pipeline para detectar locais selecionados significativamente positivos e possíveis eventos de recombinação em um alinhamento de múltiplas sequências codificadoras de proteínas. Os sites que sofrem seleção positiva fornecem insights na história evolutiva de suas seqüências, por exemplo, mostrando importantes pontos quentes da mutação, acumulados como resultados de raças de armas de vírus host durante a evolução.
Poseidon conta com uma variedade de diferentes ferramentas de terceiros (veja abaixo). Mas não se preocupe, encapsulamos cada ferramenta em seu próprio contêiner do Docker e os conectamos no sistema de gerenciamento do fluxo de trabalho NextFlow.
Vá diretamente para um pequeno exemplo de saída de Poseidon para a proteína SARS-CoV-2 em comparação com um estudo recente de Zhou et al . 2020.
Você só precisa do NextFlow (versão 20.+) e Docker instalados para executar o pipeline. Todas as dependências serão puxadas automaticamente.
Run Poseidon clonando este repositório:
clone git https://github.com/hoelzer/poseidon.gitcd Poseidon NextFlow Run Poseidon.nf -Help
ou deixe o nextflow fazer a atração
Nextflow Pull Hoelzer/Poseidon
Recomendamos usar um lançamento específico de Poseidon via
#pullNextflow Pull Hoelzer/Poseidon -r v1.0.1#runnextflow run hoelzer/Poseidon -r v1.0.1 -Help
Dependendo do seu procedimento de instalação, atualize o pipeline via git pull ou nextflow pull hoelzer/poseidon .
Importante: Poseidon precisa de sequências nucleotídicas com um quadro de leitura aberto correto como entrada. Além disso, os resultados dependem fortemente da sua seleção de seqüências, portanto, você pode considerar a execução do pipeline várias vezes com diferentes amostras de suas seqüências de entrada. Além disso, o pipeline não pode funcionar com muitas seqüências, porque em seu núcleo Poseidon usa o CodEML da suíte PAML que não é integrada para> 100 sequências. Encontre uma descrição detalhada dos parâmetros de entrada e configurações abaixo.
O NextFlow pode ser facilmente executado em diferentes ambientes, como sua máquina local, um cluster de alto desempenho ou a nuvem. Diferentes -profile são usados para informar o NextFlow qual sistema deve ser usado. Para a execução local -profile local,docker deve ser usado (e também é o padrão). Você também pode executar Poseidon em um HPC usando o singularidade via -profile lsf,singularity , -profile slurm,singularity ou -profile sge,singularity . Nesses casos, considere também ajustar --cachedir para apontar onde armazenar imagens de singularidade em seu cluster. O parâmetro --workdir também pode ser útil para ajustar onde armazenar diretórios de trabalho temporários (por exemplo, use /scratch em vez de /tmp , dependendo da sua configuração de HPC.)
Agora, vamos supor que você tenha usado o NextFlow para puxar o código Poseidon e executar o pipeline em uma máquina local usando o perfil padrão -profile local,docker .
# Mostre ajuda NextFlow Run Hoelzer/Poseidon -Help # Execute um pequeno exemplo em uma máquina local com # (primeira vez isso precisará de mais tempo porque os contêineres do docker são baixados) NextFlow Run Hoelzer/Poseidon -R v1.0.1 -Fasta ~/.Nextflow/Assets/Hoelzer/Poseidon/test_data/BATS_MX1_SMALL.FASTA -CORES 4# retomar um runnextflow broken run hoelzer/Poseidon -r v1.0.1 - -fasta ~/.nextflow/Assets/hoelzer/Poseidon/test_data/bats_mx1_small.fasta -Cores de 4 -resumo# Em vez de usar todos os núcleos disponíveis, usam apenas uma quantidade máxima no machinenextflow local run hoelzer/Poseidon -r v1.0.1 - -fasta ~/.nextflow/Assets/Hoelzer/Poseidon/test_data/Bats_mx1_small.fastA --max_cores 8 -Corores 4
Para reproduzir os resultados positivos de seleção relatados em Fuchs et al . (2017), Journal of Virology Run:
NextFlow Run Hoelzer/Poseidon -r V1.0.0 - -Fasta ~/.nextflow/Assets/Hoelzer/Poseidon/test_data/Bats_mx1.fastA -Cores 4 --kh-outgrupo "pteropus_alecto, eidolon_helvum, rousettus_aegptiacus, hypsignatus_monstrosus"-referência "myotis_daubentonii"
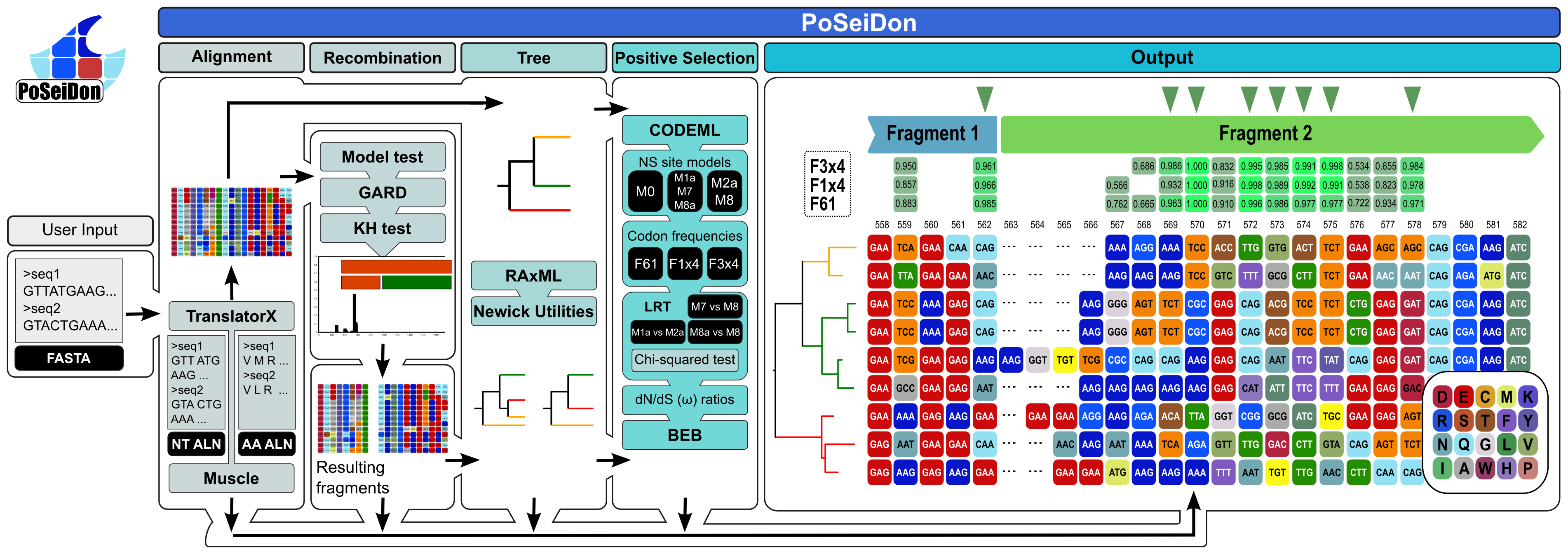
O pipeline de Poseidon compreende o alinhamento em quadro de sequências homólogas codificadoras de proteínas, detecção de eventos de recombinação putativa e pontos de interrupção evolutivos, reconstruções filogenéticas e detecção de locais selecionados positivamente no alinhamento completo e todos os fragmentos possíveis. Por fim, todos os resultados são combinados e visualizados em uma página da Web HTML amigável e fácil de usar. Os fragmentos de alinhamento resultantes são indicados com barras coloridas na saída HTML.
Translatorx (v1.1), Abascal et al . (2010); 20435676
Músculo (v3.8.31), Edgar (2004); 15034147
Raxml (v8.0.25), Stamatakis (2014); 24451623
Newick Utilities (v1.6), Junier e Zdobnov (2010); 20472542
ModelTest, Posada e Crandall (1998); 9918953
Hyphy (v2.2), Pond et al . (2005); 15509596
Gard, Pond et al. (2006); 17110367
PAML/Codeml (v4.8), Yang (2007); 17483113
Ruby (v2.3.1)
Inkscape (v1.0)
pdftex (v3.14)
A maioria dos parâmetros de Poseidon é opcional e é explicada abaixo em detalhes.
--fasta
Obrigatório. Seu arquivo fasta de entrada deve seguir o formato:
>Myotis_lucifugus Mx1 Gene ATGGCGATCGAGATACGATACGTA... >Myotis_davidii Mx1 Gene ATGGCGGTCGAGATAAGATACGTT...
Todas as seqüências devem ter um quadro de leitura aberto correto, só pode conter caracteres nucleotídeos [a | c | g | t] e nenhum códon interno de parada.
Os IDs de sequência devem ser únicos até a primeira ocorrência de um espaço.
--reference
Opcional. Padrão: use o primeiro ID da sequência como referência. Você pode definir um ID de uma espécie do seu arquivo FASTA múltiplo como uma espécie de referência. Sites selecionados positivamente e aminoácidos correspondentes serão desenhados em relação a esta espécie. O ID deve corresponder ao cabeçalho da FASTA até a ocorrência do primeiro espaço. Por exemplo, se você deseja myotis lucifugus como sua espécie de referência e seu arquivo fasta contém:
>Myotis_lucifugus Mx1 Gene ATGGCGATCGAGATACGATACGTA...
usar
--reference "Myotis_lucifugus"
como parâmetro para definir as espécies de referência. Por padrão, o primeiro ID que ocorre no arquivo múltiplo fasta será usado.
--outgroup
Opcional. Padrão: As árvores não são enraizadas. Você pode definir um ou vários IDs de espécies (separados por vírgula) como grupo externo. Todas as árvores filogenéticas estarão enraizadas de acordo com esta espécie. Por exemplo, se seu arquivo fasta múltiplo contiver
ATGGCGATCGAGATACGATACGTA...
>Myotis_davidii Mx1 Gene
ATGGCGGTCGAGATAAGATACGTT...
>Pteropus_vampyrus Mx1 Gene
ATGGCCGTAGAGATTAGATACTTT...
>Eidolon_helvum Mx1 Gene
ATGCCCGTAGAGAATAGATACTTT...você pode definir:
--outgroup "Pteropus_vampyrus,Eidolon_helvum"
enraizar todas as árvores em relação a essas duas espécies.
--kh
Opcional. Padrão: false. Com este parâmetro, você pode decidir se pontos de interrupção insignificantes devem ser levados em consideração. Todos os pontos de interrupção são testados quanto à incongruência topológica significativa usando um teste de Kashino Hasegawa (KH) Kishino, H. e Hasegawa, M. (1989). Os pontos de interrupção insignificantes de KH surgem com mais frequência da variação nos comprimentos dos ramificações entre os segmentos. No entanto, levar em consideração os pontos de interrupção insignificantes do KH pode ser interessante, porque já observamos locais putativos selecionados positivamente em fragmentos sem nenhuma incongruência topológica significativa. Os fragmentos insignificantes de KH são marcados na produção final, pois podem não ocorrer a partir de eventos reais de recombinação.
Por padrão, apenas pontos de interrupção significativos são usados para outros cálculos.
Lembre -se também de que o uso de pontos de interrupção também insignificantes pode estender o tempo de execução do Poseidon de minutos a horas, dependendo do número de pontos de interrupção detectados.
Consulte o --help para outros parâmetros (Gard, Raxml, ...) e informe -nos se você precisar de mais personalização!
Se Poseidon ajudar você a citar:
Martin Hölzer e Manja Marz, "Poseidon: um oleoduto Nextflow para a detecção de eventos de recombinação evolutiva e seleção positiva", OUP Bioinformatics (2020)


