BRAKER
v3.0.8
Aqui está uma gravação da primeira sessão do BGA23 Workshop em Braker. Se aprender a assistir a vídeos é fácil para você, considere assistir: https://www.youtube.com/watch?v=uxtkj4mukyg
Braker3 está agora em https://usegalaxy.eu/
Tsebra & Braker3 Relacionado:
Related Braker & Augustus:
Relacionado Genemark:
Mark Borodovsky, Georgia Tech, EUA, [email protected]
Tomas Bruna, Instituto Conjunto do Genoma, EUA, [email protected]
Alexandre Lomsazde, Georgia Tech, EUA, [email protected]
[A] Universidade de Greifswald, Instituto de Matemática e Ciência da Computação, Walther-Rathenau-Str. 47, 17489 Greifswald, Alemanha
[B] Universidade de Greifswald, Centro de Genômica Funcional de Micróbios, Felix-Hausdorff-Str. 8, 17489 Greifswald, Alemanha
[C] Conjunto Georgia Tech e Emory University Wallace H Coulter Departamento de Engenharia Biomédica, 30332 Atlanta, EUA
[D] Escola de Ciência e Engenharia Computacional, 30332 Atlanta, EUA
[E] Instituto de Física e Tecnologia de Moscou, Região de Moscou 141701, Dolgoprudny, Rússia
![Braker2-Team-2 [Fig10]](https://images.downcodes.com/uploads/20250214/img_67aee79a0cb7530.png)
![Braker2-Team-1 [Fig11]](https://images.downcodes.com/uploads/20250214/img_67aee79a0d1eb31.png)
![Braker2-Team-3 [Fig12]](https://images.downcodes.com/uploads/20250214/img_67aee79a0da9c32.png)
![Braker2-Team-4 [Fig13]](https://images.downcodes.com/uploads/20250214/img_67aee79a0e49933.png)
Figura 1: Autores atuais de Braker, da esquerda para a direita: Mario Stanke, Alexandre Lomsadze, Katharina J. Hoff, Tomas Bruna, Lars Gabriel e Mark Borodovsky. Reconhecemos que uma comunidade maior de cientistas contribuiu para o Código Braker (por exemplo, via solicitações de tração).
O desenvolvimento de Braker1, Braker2 e Braker3 foi apoiado pelos Institutos Nacionais de Saúde (NIH) [GM128145 para MB e MS]. O desenvolvimento do Braker3 foi parcialmente financiado pela competência de dados do projeto concedida à KJH e MS pelo governo de Mecklenburg-Vorpommern, Alemanha.
O seletor de transcrição do Braker (TSEBRA) está disponível em https://github.com/gaius-augustus/tsebra.
A GeneMark-Etp, um dos localizadores de genes no centro de Braker, está disponível em https://github.com/gatech-genemark/gegeark-etp.
Augustus, o segundo localizador de genes no centro de Braker, está disponível em https://github.com/gaius-augustus/augustus.
Galba, um spin-off de oleoduto Braker para usar o MiniProt ou o GenomeThreader para gerar genes de treinamento, está disponível em https://github.com/gaius-augustus/galba.
O número crescente de genomas sequenciados requer métodos totalmente automatizados para anotação precisa da estrutura genética. Com esse objetivo em mente, desenvolvemos o Braker1 R1 R0 , uma combinação de R2 e Augustus R3, R4 , que usa dados genômicos e RNA-seq para gerar automaticamente anotações completas da estrutura genética no novo genoma.
No entanto, a qualidade dos dados de RNA-seq que estão disponíveis para anotar um novo genoma é variável e, em alguns casos, os dados de RNA-seq não estão disponíveis.
Braker2 é uma extensão do Braker1 que permite o treinamento totalmente automatizado das ferramentas de previsão de genes Genemark-es/et/EP/ETP R14, R15, R17, F1 e Augustus de RNA-seq e/ou Informações de homologia de proteínas, e que integra o Evidências extrínsecas das informações de RNA-seq e homologia de proteínas sobre a previsão .
Em contraste com outros métodos disponíveis que dependem de informações de homologia com proteínas, o Braker2 atinge alta precisão de previsão de genes, mesmo na ausência da anotação de espécies muito intimamente relacionadas e na ausência de dados de RNA-seq.
Braker3 é o mais recente oleoduto da suíte Braker. Ele permite o uso de dados de RNA-seq e proteínas em um pipeline totalmente automatizado para treinar e prever genes altamente confiáveis com a GeneMark-Etp e Augustus. O resultado do pipeline é o conjunto de genes combinado de ambas as ferramentas de previsão de genes, que contém apenas genes com suporte muito alto a partir de evidências extrínsecas.
Neste guia do usuário, nos referiremos a Braker1, Braker2 e Braker3 simplesmente como Braker porque são executados pelo mesmo script ( braker.pl ).
Use uma montagem de genoma de alta qualidade. Se você tiver um grande número de andaimes muito curtos em sua montagem do genoma, esses andaimes curtos provavelmente aumentarão drasticamente o tempo de execução, mas não aumentarão a precisão da previsão.
Use nomes simples de andaimes no arquivo genoma (por exemplo >contig1 funcionará melhor do que >contig1my custom species namesome putative function /more/information/ and lots of special characters %&!*(){} ). Simplifique os nomes dos andaimes em todos os seus arquivos FASTA antes de executar qualquer programa de alinhamento.
Para prever genes com precisão em um novo genoma, o genoma deve ser mascarado para repetições. Isso evitará a previsão de estruturas genéticas de falsos positivos em regiões repetitivas e baixas de complexidade. O mascaramento repetido também é essencial para mapear dados de RNA-seq para um genoma com algumas ferramentas (outros mapeadores de RNA-seq, como o Hisat2, ignoram informações de mascaramento). No caso de GeneMark-es/et/EP/ETP e Augustus, o Softmasking (ou seja, colocando regiões repetidas em letras minúsculas e todas as outras regiões nas letras maiúsculas) leva a melhores resultados do que o hardmasking (ou seja, substituindo letras em regiões repetitivas pela carta N para nucleotídeo desconhecido).
Muitos genomas têm estruturas genéticas que serão previstas com precisão com parâmetros padrão de Genemark-ES/ET/EP/ETP e Augustus em Braker. No entanto, alguns genomas têm recursos específicos de clades, ou seja, o modelo de ponto de ramificação especial em fungos ou padrões não padrão do local de emenda. Leia a seção de opções [Opções] para determinar se alguma das opções personalizadas pode melhorar a precisão da previsão de genes no genoma de sua espécie -alvo.
Sempre verifique os resultados da previsão de genes antes do uso adicional! Você pode, por exemplo, usar um navegador de genoma para inspeção visual de modelos de genes em contexto com dados de evidência extrínseca. Braker suporta a geração de hubs de dados de rastreamento para o navegador do genoma do UCSC com o MakeHub para esse fim.
O Braker apresenta principalmente dados de evidência extrínseca semi-supervisionados (Informações de alinhamento RNA-seq e/ou emendado por proteínas) apoiou o treinamento de GeneMark-ES/ET/EP/ETP [F1] e o treinamento subsequente de Augustus com integração de evidências extrínsecas no final Etapa de previsão de genes. No entanto, agora existem vários dutos adicionais incluídos no Braker. A seguir, fornecemos uma visão geral dos possíveis arquivos de entrada e pipelines:
![Braker2-Main-A [Fig1]](https://images.downcodes.com/uploads/20250214/img_67aee79a0eaf534.png)
Figura 2: Braker Pipeline A: Treinando apenas a GeneMark-es em dados do genoma; previsão de genes ab initio com utensílios
![Braker2-Main-B [Fig2]](https://images.downcodes.com/uploads/20250214/img_67aee79a0f13f35.png)
Figura 3: Braker Pipeline B: Treinamento Genemark-Et suportado por informações de alinhamento emendado por RNA-seq, previsão com Augustus com as mesmas informações de alinhamento emendado.
![Braker2-Main-C [Fig3]](https://images.downcodes.com/uploads/20250214/img_67aee79a0fa1036.png)
Figura 4: Braker Pipeline C: Treinando Genemark-EP+ sobre alinhamento emendado por proteínas, Informações de início e parada, previsão com Augustus com a mesma informação, além de dicas de cdspart acorrentas. As proteínas usadas aqui podem ser de qualquer distância evolutiva do organismo -alvo.
![Braker3-Main-A [Fig4]](https://images.downcodes.com/uploads/20250214/img_67aee79a1010b37.png)
Figura 5: Braker Pipeline D: Se necessário, faça o download e o alinhamento dos conjuntos de RNA-seq para as espécies-alvo. Treinamento de Genemark-ETP suportado pelos alinhamentos RNA-seq e um grande banco de dados de proteínas (as proteínas podem ser de qualquer distância evolutiva). Posteriormente, o treinamento e a previsão de Augustus usando as mesmas informações extrínsecas juntamente com os resultados da GeneMark-ETP. A previsão final é a combinação TSEBRA dos resultados de Augusto e GeneMark-ETP.
Estamos cientes de que a instalação "manual" do Braker3 e todas as suas dependências é tediosa e realmente desafiadora sem permissões de raiz. Portanto, fornecemos um recipiente do docker que foi desenvolvido para ser executado com singularidade. Todas as informações sobre este contêiner podem ser encontradas em https://hub.docker.com/r/teambraker/braker3
Em suma, construa o seguinte:
singularity build braker3.sif docker://teambraker/braker3:latest
Executar com:
singularity exec braker3.sif braker.pl
Teste com:
singularity exec -B $PWD:$PWD braker3.sif cp /opt/BRAKER/example/singularity-tests/test1.sh .
singularity exec -B $PWD:$PWD braker3.sif cp /opt/BRAKER/example/singularity-tests/test2.sh .
singularity exec -B $PWD:$PWD braker3.sif cp /opt/BRAKER/example/singularity-tests/test3.sh .
export BRAKER_SIF=/your/path/to/braker3.sif # may need to modify
bash test1.sh
bash test2.sh
bash test3.sh
Poucos usuários desejam executar sua análise dentro do Docker (pois são necessárias permissões de raiz). No entanto, se esse é o seu objetivo, você pode executar e testar o contêiner como segue
sudo docker run --user 1000:100 --rm -it teambraker/braker3:latest bash
bash /opt/BRAKER/example/docker-tests/test1.sh # BRAKER1
bash /opt/BRAKER/example/docker-tests/test2.sh # BRAKER2
bash /opt/BRAKER/example/docker-tests/test3.sh # BRAKER3
Boa sorte ;-)
$PATH podem levar a interferências imprevistas, causando falhas no programa. Por favor, mova todas as versões da GeneMark mais antigas do seu $PATH (também, por exemplo, o Genemark em ProtHint/dependencies ).
No momento do lançamento, esta versão do Braker foi testada com:
Augustus 3.5.0 F2
Genemark-Etp (fonte ver Dockerfile)
Bamtools 2.5.1 R5
Samtools 1.7-4-G93586ed R6
Spaln 2.3.3d R8, R9, R10
NCBI BLAST+ 2.2.31+ R12, R13
Diamante 0.9.24
CDBFastA 0,99
CDBYANK 0,981
Gushr 1.0.0
SRA Toolkit 3.00 R14
Hisat2 2.2.1 R15
Bedtools 2.30 R16
Stringtie2 2.2.1 R17
GFFREAD 0.12,7 R18
Completo 0.2.5 R27
A corrida de Braker requer um sistema Linux com bash e Perl. Além disso, Braker exige que os seguintes módulos CPAN-PERL sejam instalados:
File::Spec::Functions
Hash::Merge
List::Util
MCE::Mutex
Module::Load::Conditional
Parallel::ForkManager
POSIX
Scalar::Util::Numeric
YAML
Math::Utils
File::HomeDir
Para a GeneMark-Etp, usada quando a proteína e o RNA-seq são fornecidos:
YAML::XSData::DumperThread::Queuethreads No Ubuntu, por exemplo, instale os módulos com o módulo cpanminminus f4 : sudo cpanm Module::Name , por exemplo, sudo cpanm Hash::Merge .
Braker também usa um módulo Perl helpMod_braker.pm que não está disponível no CPAN. Este módulo faz parte da versão do Braker e não requer instalação separada.
Se você não tiver permissões de raiz na máquina Linux, tente configurar um ambiente Anaconda (https://www.anaconda.com/distribution/) da seguinte maneira:
wget https://repo.anaconda.com/archive/Anaconda3-2018.12-Linux-x86_64.sh
bash bin/Anaconda3-2018.12-Linux-x86_64.sh # do not install VS (needs root privileges)
conda install -c anaconda perl
conda install -c anaconda biopython
conda install -c bioconda perl-app-cpanminus
conda install -c bioconda perl-file-spec
conda install -c bioconda perl-hash-merge
conda install -c bioconda perl-list-util
conda install -c bioconda perl-module-load-conditional
conda install -c bioconda perl-posix
conda install -c bioconda perl-file-homedir
conda install -c bioconda perl-parallel-forkmanager
conda install -c bioconda perl-scalar-util-numeric
conda install -c bioconda perl-yaml
conda install -c bioconda perl-class-data-inheritable
conda install -c bioconda perl-exception-class
conda install -c bioconda perl-test-pod
conda install -c bioconda perl-file-which # skip if you are not comparing to reference annotation
conda install -c bioconda perl-mce
conda install -c bioconda perl-threaded
conda install -c bioconda perl-list-util
conda install -c bioconda perl-math-utils
conda install -c bioconda cdbtools
conda install -c eumetsat perl-yaml-xs
conda install -c bioconda perl-data-dumper
Posteriormente, instale o Braker e outros softwares "como de costume" enquanto estiver em seu ambiente de conda. Nota: Existe um pacote Bioconda Braker e um pacote Bioconda Augustus. Eles funcionam. Mas eles geralmente estão atrasados para o código de desenvolvimento de ambas as ferramentas no GitHub. Portanto, recomendamos a instalação manual e o uso de fontes mais recentes.
Braker é uma coleção de scripts Perl e Python e um módulo Perl. O script principal que será chamado para executar o Braker é braker.pl . Os componentes adicionais Perl e Python são:
align2hints.pl
filterGenemark.pl
filterIntronsFindStrand.pl
startAlign.pl
helpMod_braker.pm
findGenesInIntrons.pl
downsample_traingenes.pl
ensure_n_training_genes.py
get_gc_content.py
get_etp_hints.py
Todos os scripts (arquivos que terminam com *.pl e *.py ) que fazem parte do Braker devem ser executáveis para executar o Braker. Já deve ser o caso se você baixar o Braker do Github. A executabilidade pode ser substituída se você, por exemplo, transferir o Braker em um stick USB para outro computador. Para verificar se os arquivos necessários são executáveis, execute o seguinte comando no diretório que contém scripts Braker Perl:
ls -l *.pl *.py
A saída deve ser semelhante a este:
-rwxr-xr-x 1 katharina katharina 18191 Mai 7 10:25 align2hints.pl
-rwxr-xr-x 1 katharina katharina 6090 Feb 19 09:35 braker_cleanup.pl
-rwxr-xr-x 1 katharina katharina 408782 Aug 17 18:24 braker.pl
-rwxr-xr-x 1 katharina katharina 5024 Mai 7 10:25 downsample_traingenes.pl
-rwxr-xr-x 1 katharina katharina 5024 Mai 7 10:23 ensure_n_training_genes.py
-rwxr-xr-x 1 katharina katharina 4542 Apr 3 2019 filter_augustus_gff.pl
-rwxr-xr-x 1 katharina katharina 30453 Mai 7 10:25 filterGenemark.pl
-rwxr-xr-x 1 katharina katharina 5754 Mai 7 10:25 filterIntronsFindStrand.pl
-rwxr-xr-x 1 katharina katharina 7765 Mai 7 10:25 findGenesInIntrons.pl
-rwxr-xr-x 1 katharina katharina 1664 Feb 12 2019 gatech_pmp2hints.pl
-rwxr-xr-x 1 katharina katharina 2250 Jan 9 13:55 log_reg_prothints.pl
-rwxr-xr-x 1 katharina katharina 4679 Jan 9 13:55 merge_transcript_sets.pl
-rwxr-xr-x 1 katharina katharina 41674 Mai 7 10:25 startAlign.pl
É importante que o x in -rwxr-xr-x esteja presente para cada script. Se não for esse o caso, corra
`chmod a+x *.pl *.py`
Para alterar os atributos do arquivo.
Você pode achar útil adicionar o diretório no qual os scripts do Braker Perl residem à sua variável de ambiente $PATH . Para uma única sessão de Bash, digite:
PATH=/your_path_to_braker/:$PATH
export PATH
Para disponibilizar essa modificação $PATH para todas as sessões de bash, adicione as linhas acima a um script de inicialização (por exemplo ~/.bashrc ).
Braker pede várias ferramentas de software de bioinformática que não fazem parte do Braker. Algumas ferramentas são obrigatórias, o IE Braker não será executado se essas ferramentas não estiverem presentes no seu sistema. Outras ferramentas são opcionais. Instale todas as ferramentas necessárias para a execução do Braker no modo de sua escolha.
Faça o download da GeneMark-Etp F1 em http://github.com/gatech-genemark/gegeark-etp ou https://topaz.gatech.edu/genemark/etp.for_braker.tar.gz. Desembore e instale o GeneMark-Etp, conforme descrito no arquivo README da GeneMark-Etp.
Se já contido na sua variável $PATH , o Braker adivinhará a localização do gmes_petap.pl ou gmetp.pl automaticamente. Caso contrário, o Braker pode encontrar os executáveis Genemark-es/ET/EP/ETP, localizando-os em uma variável de ambiente GENEMARK_PATH ou assumindo um argumento da linha de comando ( --GENEMARK_PATH=/your_path_to_GeneMark_executables/ ).
Para definir a variável de ambiente para a sua sessão de bash atual, digite:
export GENEMARK_PATH=/your_path_to_GeneMark_executables/
Adicione as linhas acima a um script de inicialização (por exemplo, ~/.bashrc ) para disponibilizá -lo para todas as sessões da Bash.
Os scripts Perl no GeneMark-ES/ET/EP/ETP são configurados com a localização PERL padrão em /usr/bin/perl .
Se você estiver executando a GeneMark-es/et/EP/ETP em um ambiente da Anaconda (ou deseja usar o Perl da variável $PATH por qualquer outro motivo), modifique o shebang de todos os scripts Genemark-es/et/EP/ETP com O comando a seguir localizado dentro da pasta GeneMark-es/et/EP/ETP:
perl change_path_in_perl_scripts.pl "/usr/bin/env perl"
Você pode verificar se o Genemark-es/ET/EP está instalado corretamente executando o check_install.bash e/ou executando exemplos no diretório GeneMark-E-tests .
A GeneMark-Etp é compatível descendente, ou seja, também abrange a funcionalidade do Genemark-Ep e Genemark-et em Braker.
Faça o download de Augustus de sua filial mestre em https://github.com/gaius-augustus/augustus. Desembale Augustus e instale Augustus de acordo com Augustus README.TXT . Não use versões de Augustus desatualizadas de outras fontes, por exemplo, pacote Debian ou pacote bioconda! Braker depende muito, em particular, de um diretório atualizado de Augustus/Scripts, e outras fontes geralmente ficam para trás.
Você deve compilar Augustus em seu próprio sistema para evitar problemas com versões de bibliotecas usadas por Augustus. As instruções de compilação são fornecidas no arquivo Augustus README.TXT ( Augustus/README.txt ).
Augustus consiste em augustus , a ferramenta de previsão de genes, ferramentas de C ++ adicionais localizadas nos scripts Augustus/auxprogs e Perl localizados em Augustus/scripts . Os scripts Perl devem ser executados (consulte as instruções nos componentes da seção Braker.
A ferramenta C ++ bam2hints é um componente essencial do Braker quando executado com RNA-seq. As fontes estão localizadas em Augustus/auxprogs/bam2hints . Certifique -se de compilar bam2hints no seu sistema (ele deve ser compilado automaticamente quando Augustus for compilado, mas em caso de problemas com bam2hints , leia instruções de solução de problemas em Augustus/auxprogs/bam2hints/README ).
Como Braker é um pipeline que treina Augustus, ou seja, escreve arquivos de parâmetros específicos de espécies, Braker precisa escrever acesso ao diretório de configuração de Augustus que contém esses arquivos ( Augustus/config/ ). Se você instalar Augustus globalmente no seu sistema, a pasta config normalmente não será gravada por todos os usuários. Faça o diretório em que config reside recursivamente gravável para os usuários de Augustus ou copie a config/ pasta (recursivamente) para um local onde os usuários têm permissão de escrita.
Augustus localizará a pasta config procurando uma variável de ambiente $AUGUSTUS_CONFIG_PATH . Se a variável de ambiente $AUGUSTUS_CONFIG_PATH não estiver definida, Braker procurará no caminho ../config em relação ao diretório em que encontra um executável de Augustus. Como alternativa, você pode fornecer a variável como um argumento da linha de comando para Braker ( --AUGUSTUS_CONFIG_PATH=/your_path_to_AUGUSTUS/Augustus/config/ ). Recomendamos que você exporte a variável, por exemplo, para sua sessão atual de bash:
export AUGUSTUS_CONFIG_PATH=/your_path_to_AUGUSTUS/Augustus/config/
Para disponibilizar a variável para todas as sessões de bash, adicione a linha acima a um script de inicialização, por exemplo ~/.bashrc .
Por favor, dê uma olhada no Dockerfile, caso você queira instalar Augustus como pacote Debian. Vários scripts precisam ser corrigidos, então.
Braker espera que todo o diretório config de Augustus, em $AUGUSTUS_CONFIG_PATH , ou seja, as species das subpastas com seu conteúdo (pelo menos generic ) e extrinsic ! Fornecer uma pasta gravável, mas vazia em $AUGUSTUS_CONFIG_PATH não funcionará para Braker. Se você precisar separar o Augustus Binary e $AUGUSTUS_CONFIG_PATH , recomendamos que você copie recursivamente o conteúdo de configuração não criativo para um local gravável.
Se você possui uma instalação em todo o sistema de Augustus em /usr/bin/augustus , uma cópia do config fica em /usr/bin/augustus_config/ . A pasta /home/yours/ é gravável para você. Copie com o seguinte comando (e defina as variáveis então necessárias):
cp -r /usr/bin/Augustus/config/ /home/yours/
export AUGUSTUS_CONFIG_PATH=/home/yours/augustus_config
export AUGUSTUS_BIN_PATH=/usr/bin
export AUGUSTUS_SCRIPTS_PATH=/usr/bin/augustus_scripts
A adição de diretórios de binários e scripts de Augustus à sua variável $PATH permite que seu sistema localize essas ferramentas automaticamente. Não é um requisito para a corrida de Braker fazer isso, porque Braker tentará adivinhá -los a partir da localização de outra variável de ambiente ( $AUGUSTUS_CONFIG_PATH ), ou ambos os diretórios podem ser fornecidos como argumentos da linha de comando para braker.pl , mas recomendamos Adicione -os à sua variável $PATH . Para a sua sessão atual, digite:
PATH=:/your_path_to_augustus/bin/:/your_path_to_augustus/scripts/:$PATH
export PATH
Para todas as suas sessões bash, adicione as linhas acima a um script de inicialização (por exemplo ~/.bashrc ).
No Ubuntu, o Python3 geralmente é instalado por padrão, python3 estará na sua variável $PATH , por padrão, e o Braker o localizará automaticamente. No entanto, você tem a opção de especificar o local binário python3 de duas outras maneiras:
Exportar uma variável de ambiente $PYTHON3_PATH , por exemplo, seu arquivo ~/.bashrc :
export PYTHON3_PATH=/path/to/python3/
Especifique a opção de linha de comando --PYTHON3_PATH=/path/to/python3/ to braker.pl .
Faça o download do Bamtools (por exemplo, git clone https://github.com/pezmaster31/bamtools.git ). Instale os Bamtools digitando o seguinte em seu shell:
cd your-bamtools-directory mkdir build cd build cmake .. make
Se já estiver na sua variável $PATH , o Braker encontrará Bamtools, automaticamente. Caso contrário, o Braker pode localizar o binário do Bamtools usando uma variável de ambiente $BAMTOOLS_PATH ou pegando um argumento da linha de comando ( --BAMTOOLS_PATH=/your_path_to_bamtools/bin/ f6 ). Para definir a variável de ambiente, por exemplo, para sua sessão de bash atual, digite:
export BAMTOOLS_PATH=/your_path_to_bamtools/bin/
Adicione a linha acima a um script de inicialização (por exemplo ~/.bashrc ) para definir a variável de ambiente para todas as sessões de bash.
Você pode usar o NCBI BLAST+ ou o diamante para remover genes de treinamento redundantes. Você não precisa de ambas as ferramentas. Se o diamante estiver presente, será preferido porque é muito mais rápido.
Obtenha e descompacte o diamante da seguinte forma:
wget http://github.com/bbuchfink/diamond/releases/download/v0.9.24/diamond-linux64.tar.gz
tar xzf diamond-linux64.tar.gz
Se já estiver na sua variável $PATH , o Braker encontrará diamante, automaticamente. Caso contrário, o Braker pode localizar o binário de diamante usando uma variável de ambiente $DIAMOND_PATH ou assumindo um argumento da linha de comando ( --DIAMOND_PATH=/your_path_to_diamond ). Para definir a variável de ambiente, por exemplo, para sua sessão de bash atual, digite:
export DIAMOND_PATH=/your_path_to_diamond/
Adicione a linha acima a um script de inicialização (por exemplo ~/.bashrc ) para definir a variável de ambiente para todas as sessões de bash.
Se você decidir por BLAST+, instale o NCBI BLAST+ com sudo apt-get install ncbi-blast+ .
Se já estiver em sua variável $PATH , o Braker encontrará BLASTP, automaticamente. Caso contrário, o Braker pode localizar o binário blastp usando uma variável de ambiente $BLAST_PATH ou assumindo um argumento da linha de comando ( --BLAST_PATH=/your_path_to_blast/ ). Para definir a variável de ambiente, por exemplo, para sua sessão de bash atual, digite:
export BLAST_PATH=/your_path_to_blast/
Adicione a linha acima a um script de inicialização (por exemplo ~/.bashrc ) para definir a variável de ambiente para todas as sessões de bash.
As ferramentas a seguir são necessárias pelo GeneMark-Etp e tentará localizá-las na sua variável $PATH . Portanto, adicione sua localização ao seu $PATH , por exemplo:
export PATH=$PATH:/your/path/to/Tool
Para todas as ferramentas abaixo, adicione a linha acima a um script de inicialização $PATH por exemplo ~/.bashrc
Essas ferramentas de software são apenas obrigatórias se você executar o Braker com dados de RNA-seq e proteínas!
Stringtie2 é usado pelo GeneMark-Etp para montar alinhamentos alinhados de RNA-seq. Uma versão pré -compilada do Stringtie2 pode ser baixada em https://ccb.jhu.edu/software/stringtie/#install.
O pacote de software Bedtools é necessário pelo GeneMark-Etp se você deseja executar o Braker com dados de RNA-seq e proteína. Você pode baixar o Bedtools em https://github.com/arq5x/bedtools2/releases. Aqui, você pode baixar uma versão pré -compilada bedtools.static.binary , por exemplo
wget https://github.com/arq5x/bedtools2/releases/download/v2.30.0/bedtools.static.binary
mv bedtools.static.binary bedtools
chmod a+x
Ou você pode baixar bedtools-2.30.0.tar.gz e compilá-lo da fonte usando make , por exemplo
wget https://github.com/arq5x/bedtools2/releases/download/v2.30.0/bedtools-2.30.0.tar.gz
tar -zxvf bedtools-2.30.0.tar.gz
cd bedtools2
make
Consulte https://bedtools.readthedocs.io/en/latest/content/installation.html para obter mais informações.
O GFFREAD é um software de utilitário exigido pelo GeneMark-Etp. Ele pode ser baixado em https://github.com/gpertea/gffread/releases/download/v0.12.7/gffread-0.12.7.linux_x86_64.tar.gz e instalado com make , por exemplo
wget https://github.com/gpertea/gffread/releases/download/v0.12.7/gffread-0.12.7.Linux_x86_64.tar.gz
tar xzf gffread-0.12.7.Linux_x86_64.tar.gz
cd gffread-0.12.7.Linux_x86_64
make
O Samtools não é necessário para a execução do Braker sem a GeneMark-Etp se todos os seus arquivos forem formatados, corretamente (ou seja, todas as seqüências devem ter nomes de fasta curtos e únicos). Se você não tiver certeza se todos os seus arquivos são fomatados corretamente, pode ser útil instalar o Samtools porque o Braker pode corrigir automaticamente determinados problemas de formato usando o Samtools.
Como um pré -requisito para o Samtools, faça o download e instale htslib (por exemplo, git clone https://github.com/samtools/htslib.git , siga a documentação htslib para instalação).
Faça o download e instale o samtools (por exemplo git clone git://github.com/samtools/samtools.git ), siga posteriormente a documentação do Samtools para instalação).
Se já estiver em sua variável $PATH , o Braker encontrará o Samtools, automaticamente. Caso contrário, o Braker pode encontrar o Samtools assumindo um argumento da linha de comando ( --SAMTOOLS_PATH=/your_path_to_samtools/ ) ou usando uma variável de ambiente $SAMTOOLS_PATH . Para exportar a variável, por exemplo, para sua sessão de batida atual, digite:
export SAMTOOLS_PATH=/your_path_to_samtools/
Adicione a linha acima a um script de inicialização (por exemplo ~/.bashrc ) para definir a variável de ambiente para todas as sessões de bash.
Se o BiopyThon estiver instalado, o Braker poderá gerar arquivos de fasta com sequências de codificação e seqüências de proteínas previstas por Augustus e gerar cubos de dados de trilha para visualização de um Braker Run com o MakeHub R16 . Estas são etapas opcionais. O primeiro pode ser desativado com o sinalizador da linha de comando --skipGetAnnoFromFasta , o segundo pode ser ativado usando as opções da linha de comando --makehub [email protected] , biopython não for necessário se nenhuma dessas etapas opcionais deve ser realizado.
No Ubuntu, instale o Python3 Package Manager com:
`sudo apt-get install python3-pip`
Em seguida, instale Biopython com:
`sudo pip3 install biopython`
CDBFastA e CDByank são exigidos pelo Braker para corrigir os genes de Augustus com códons de parada no quadro (códons de parada emendados) usando o script Augustus FIX_IN_FRAM_STOP_CODON_GENES.PY. Isso pode ser ignorado com --skip_fixing_broken_genes .
No Ubuntu, instale o CDBFastA com:
sudo apt-get install cdbfasta
Para outros sistemas, você pode, por exemplo, obter CDBFastA em https://github.com/gpertea/cdbfasta, por exemplo:
git clone https://github.com/gpertea/cdbfasta.git
cd cdbfasta
make all
No Ubuntu, o CDBFastA e o CDByank estarão na sua variável $PATH após a instalação, e o Braker os localizará automaticamente. No entanto, você tem a opção de especificar a localização binária cdbfasta e cdbyank de duas outras maneiras:
$CDBTOOLS_PATH , por exemplo, no seu arquivo ~/.bashrc : export CDBTOOLS_PATH=/path/to/cdbtools/
--CDBTOOLS_PATH=/path/to/cdbtools/ TO braker.pl . NOTA: O apoio ao Spaln independente (ouside de Prothint) em Braker é descontinuado.
Essa ferramenta é necessária se você executar o ProThint ou se deseja executar alinhamentos de proteínas para genoma com Braker usando Spaln fora do Prothint. Usar Spaln fora do Prothint é uma abordagem adequada apenas se uma espécie anotada de curta distância evolutiva do seu genoma alvo estiver disponível. Recomendamos correr Spaln através do Prothint para Braker. Prothint traz um binário de Spaln. Se isso não funcionar no seu sistema, faça o download do spaln em https://github.com/ogotoh/spaln. Desembore e instale de acordo com spaln/doc/SpalnReadMe22.pdf .
Braker tentará localizar o executável SPALN usando uma variável de ambiente $ALIGNMENT_TOOL_PATH . Como alternativa, isso pode ser fornecido como argumento da linha de comando ( --ALIGNMENT_TOOL_PATH=/your/path/to/spaln ).
Essa ferramenta é necessária apenas se você deseja adicionar UTRs (dos dados RNA-seq) a genes previstos ou se você deseja treinar parâmetros da UTR para Augustus e prever genes com UTRs. De qualquer forma, o Gushr requer a entrada de dados de RNA-seq.
Gushr está disponível para download em https://github.com/gaius-augustus/gushr. Obtê -lo digitando:
git clone https://github.com/Gaius-Augustus/GUSHR.git
Gushr executa um arquivo JAR de Gemoma R19, R20, R21 , e este arquivo JAR requer Java 1.8. No Ubuntu, você pode instalar o Java 1.8 com o seguinte comando:
sudo apt-get install openjdk-8-jdk
Se você possui várias versões Java instaladas em seu sistema, certifique -se de ativar 1,8 Braker anterior com Java, executando
sudo update-alternatives --config java
e selecionando a versão correta.
Se você alternar --UTR=on , o Bamtowig.py exigirá as seguintes ferramentas que podem ser baixadas em http://hgdownload.soe.ucsc.edu/admin/exe:
Twobitinfo
Fatotwobit
É opcional instalar essas ferramentas no seu $ PATH. Se não o fizer, e você alterna --UTR=on , o Bamtowig.py os baixará automaticamente no diretório de trabalho.
Se você deseja automaticamente, gerar um hub de dados da sua execução de Braker, o software MakeHub, disponível em https://github.com/gaius-augustus/makehub. Baixe o software (executando git clone https://github.com/Gaius-Augustus/MakeHub.git , ou escolhendo um lançamento em https://github.com/gaius-augustus/makehub/releases. Extract the Release Pacote se você baixou um lançamento (por exemplo, unzip MakeHub.zip ou tar -zxvf MakeHub.tar.gz .
Braker tentará localizar o script make_hub.py usando uma variável de ambiente $MAKEHUB_PATH . Como alternativa, isso pode ser fornecido como argumento da linha de comando ( --MAKEHUB_PATH=/your/path/to/MakeHub/ ). Braker também pode tentar adivinhar a localização do MakeHub no seu sistema.
Se você deseja que o Braker baixe as bibliotecas RNA-seq do SRA do NCBI, o SRA Toolkit é necessário. Você pode obter uma versão pré-compilada do SRA Toolkit de http://daehwankimlab.github.io/hisat2/download/#version-hisat2-221.
Braker tentará encontrar binários executáveis do SRA Toolkit (FastQ-DUMP, PREFICT) usando uma variável de ambiente $SRATOOLS_PATH . Como alternativa, isso pode ser fornecido como argumento da linha de comando ( --SRATOOLS_PATH=/your/path/to/SRAToolkit/ ). O Braker também pode tentar adivinhar a localização do SRA Toolkit no seu sistema se os executáveis estiverem na sua variável $PATH .
Se você deseja usar leituras de RNA-seq inalignadas, o software Hisat2 é necessário para mapeá-los para o genoma. Uma versão pré-compilada do Hisat2 pode ser baixada em http://daehwankimlab.github.io/hisat2/download/#version-hisat2-221.
Braker tentará encontrar binários executáveis HISAT2 (Hisat2, Hisat2-Build) usando uma variável de ambiente $HISAT2_PATH . Como alternativa, isso pode ser fornecido como argumento da linha de comando ( --HISAT2_PATH=/your/path/to/HISAT2/ ). O Braker também pode tentar adivinhar a localização do Hisat2 em seu sistema se os executáveis estiverem na sua variável $PATH .
Se você deseja executar o TSEBRA em Braker em um modo de maximização da integridade do Busco, precisará instalar o CompleASM.
wget https://github.com/huangnengCSU/compleasm/releases/download/v0.2.4/compleasm-0.2.4_x64-linux.tar.bz2
tar -xvjf compleasm-0.2.4_x64-linux.tar.bz2 &&
Adicione a pasta resultante compliqueasm_kit à sua variável $PATH , por exemplo:
export PATH=$PATH:/your/path/to/compleasm_kit
CompleASM requer pandas, que podem ser instalados com:
pip install pandas
Braker (Braker.pl) usa o getConf para ver quantos threads podem ser executados no seu sistema. No Ubuntu, você pode instalá -lo com:
sudo apt-get install libc-bin
A seguir, descrevemos o Braker "típico" exige diferentes tipos de dados de entrada. Em geral, recomendamos que você execute Braker em seqüências genômicas que foram máscaras soft para repetições. Braker deve ser aplicado apenas a genomas que foram máscaras soft para repetições!
This approach is suitable for genomes of species for which RNA-Seq libraries with good transcriptome coverage are available and for which protein data is not at hand. The pipeline is illustrated in Figure 2.
BRAKER has several ways to receive RNA-Seq data as input:
You can provide ID(s) of RNA-Seq libraries from SRA (in case of multiple IDs, separate them by comma) as argument to --rnaseq_sets_ids . The libraries belonging to the IDs are then downloaded automatically by BRAKER, eg:
braker.pl --species=yourSpecies --genome=genome.fasta
--rnaseq_sets_ids=SRA_ID1,SRA_ID2
You can use local FASTQ file(s) of unaligned reads as input. In this case, you have to provide BRAKER with the ID(s) of the RNA-Seq set(s) as argument to --rnaseq_sets_ids and the path(s) to the directories, where the FASTQ files are located as argument to --rnaseq_sets_dirs . For each ID ID , BRAKER will search in these directories for one FASTQ file named ID.fastq if the reads are unpaired, or for two FASTQ files named ID_1.fastq and ID_2.fastq if they are paired.
For example, if you have a paired library called 'SRA_ID1' and an unpaired library named 'SRA_ID2', you have to have a directory /path/to/local/fastq/files/ , where the files SRA_ID1_1.fastq , SRA_ID1_2.fastq , and SRA_ID2.fastq reside. Then, you could run BRAKER with following command:
braker.pl --species=yourSpecies --genome=genome.fasta
--rnaseq_sets_ids=SRA_ID1,SRA_ID2
--rnaseq_sets_dirs=/path/to/local/fastq/files/
There are two ways of supplying BRAKER with RNA-Seq data as bam file(s). First, you can do it in the same way as you would supply FASTQ file(s): Provide the ID(s)/name(s) of your bam file(s) as argument to --rnaseq_sets_ids and specify directories where the bam files reside with --rnaseq_sets_dirs . BRAKER will automatically detect that these ID(s) are bam and not FASTQ file(s), eg:
braker.pl --species=yourSpecies --genome=genome.fasta
--rnaseq_sets_ids=BAM_ID1,BAM_ID2
--rnaseq_sets_dirs=/path/to/local/bam/files/
Second, you can specify the paths to your bam file(s) directly, eg can either extract RNA-Seq spliced alignment information from bam files, or it can use such extracted information, directly.
braker.pl --species=yourSpecies --genome=genome.fasta
--bam=file1.bam,file2.bam
Please note that we generally assume that bam files were generated with HiSat2 because that is the aligner that would also be executed by BRAKER3 with fastq input. If you want for some reason to generate the bam files with STAR, use the option --outSAMstrandField intronMotif of STAR to produce files that are compatible wiht StringTie in BRAKER3.
In order to run BRAKER with RNA-Seq spliced alignment information that has already been extracted, run:
braker.pl --species=yourSpecies --genome=genome.fasta
--hints=hints1.gff,hints2.gff
The format of such a hints file must be as follows (tabulator separated file):
chrName b2h intron 6591 8003 1 + . pri=4;src=E
chrName b2h intron 6136 9084 11 + . mult=11;pri=4;src=E
...
The source b2h in the second column and the source tag src=E in the last column are essential for BRAKER to determine whether a hint has been generated from RNA-Seq data.
It is also possible to provide RNA-Seq sets in different ways for the same BRAKER run, any combination of above options is possible. It is not recommended to provide RNA-Seq data with --hints if you run BRAKER in ETPmode (RNA-Seq and protein data), because GeneMark-ETP won't use these hints!
This approach is suitable for genomes of species for which no RNA-Seq libraries are available. A large database of proteins (with possibly longer evolutionary distance to the target species) should be used in this case. This mode is illustrated in figure 9.
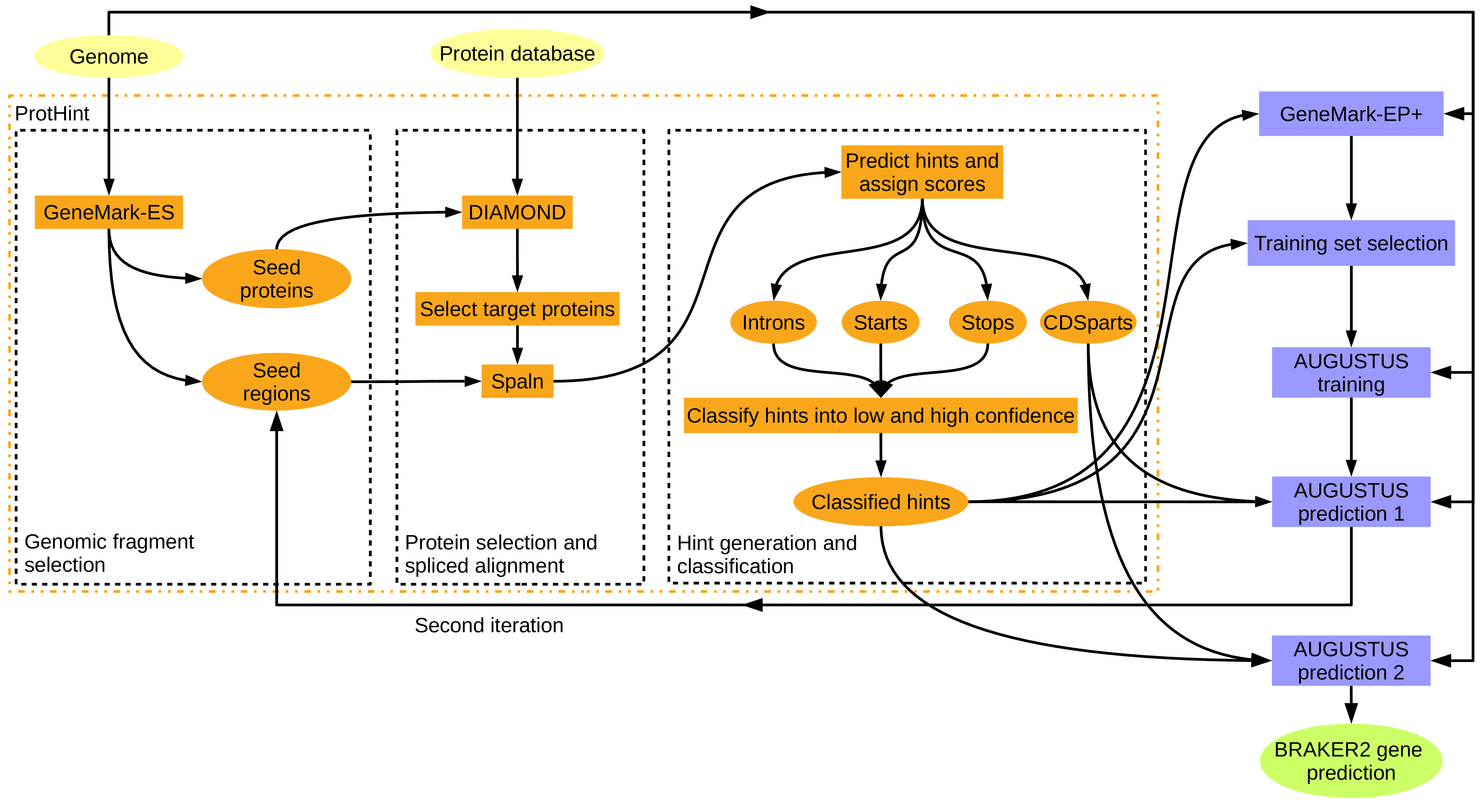
Figure 9: BRAKER with proteins of any evolutionary distance. ProtHint protein mapping pipelines is used to generate protein hints. ProtHint automatically determines which alignments are from close relatives, and which are from rather distant relatives.
For running BRAKER in this mode, type:
braker.pl --genome=genome.fa --prot_seq=proteins.fa
We recommend using OrthoDB as basis for proteins.fa . The instructions on how to prepare the input OrthoDB proteins are documented here: https://github.com/gatech-genemark/ProtHint#protein-database-preparation.
You can of course add additional protein sequences to that file, or try with a completely different database. Any database will need several representatives for each protein, though.
Instead of having BRAKER run ProtHint, you can also start BRAKER with hints already produced by ProtHint, by providing ProtHint's prothint_augustus.gff output:
braker.pl --genome=genome.fa --hints=prothint_augustus.gff
The format of prothint_augustus.gff in this mode looks like this:
2R ProtHint intron 11506230 11506648 4 + . src=M;mult=4;pri=4
2R ProtHint intron 9563406 9563473 1 + . grp=69004_0:001de1_702_g;src=C;pri=4;
2R ProtHint intron 8446312 8446371 1 + . grp=43151_0:001cae_473_g;src=C;pri=4;
2R ProtHint intron 8011796 8011865 2 - . src=P;mult=1;pri=4;al_score=0.12;
2R ProtHint start 234524 234526 1 + . src=P;mult=1;pri=4;al_score=0.08;
The prediction of all hints with src=M will be enforced. Hints with src=C are 'chained evidence', ie they will only be incorporated if all members of the group (grp=...) can be incorporated in a single transcript. All other hints have src=P in the last column. Supported features in column 3 are intron , start , stop and CDSpart .
If RNA-Seq (and only RNA-Seq) data is provided to BRAKER as a bam-file, and if the genome is softmasked for repeats, BRAKER can automatically train UTR parameters for AUGUSTUS. After successful training of UTR parameters, BRAKER will automatically predict genes including coverage information form RNA-Seq data. Example call:
braker.pl --species=yourSpecies --genome=genome.fasta
--bam=file.bam --UTR=on
Warnings:
This feature is experimental!
--UTR=on is currently not compatible with bamToWig.py as released in AUGUSTUS 3.3.3; it requires the current development code version from the github repository (git clone https://github.com/Gaius-Augustus/Augustus.git).
--UTR=on increases memory consumption of AUGUSTUS. Carefully monitor jobs if your machine was close to maxing RAM without --UTR=on! Reducing the number of cores will also reduce RAM consumption.
UTR prediction sometimes improves coding sequence prediction accuracy, but not always. If you try this feature, carefully compare results with and without UTR parameters, afterwards (eg in UCSC Genome Browser).
For running BRAKER without UTR parameters, it is not very important whether RNA-Seq data was generated by a stranded protocol (because spliced alignments are 'artificially stranded' by checking the splice site pattern). However, for UTR training and prediction, stranded libraries may provide information that is valuable for BRAKER.
After alignment of the stranded RNA-Seq libraries, separate the resulting bam file entries into two files: one for plus strand mappings, one for minus strand mappings. Call BRAKER as follows:
braker.pl --species=yourSpecies --genome=genome.fasta
--bam=plus.bam,minus.bam --stranded=+,-
--UTR=on
You may additionally include bam files from unstranded libraries. Those files will not used for generating UTR training examples, but they will be included in the final gene prediction step as unstranded coverage information, example call:
braker.pl --species=yourSpecies --genome=genome.fasta
--bam=plus.bam,minus.bam,unstranded.bam
--stranded=+,-,. --UTR=on
Warning: This feature is experimental and currently has low priority on our maintenance list!
The native mode for running BRAKER with RNA-Seq and protein data. This will call GeneMark-ETP, which will use RNA-Seq and protein hints for training GeneMark-ETP. Subsequently, AUGUSTUS is trained on 'high-confindent' genes (genes with very high extrinsic evidence support) from the GeneMark-ETP prediction and a set of genes is predicted by AUGUSTUS. In a last step, the predictions of AUGUSTUS and GeneMark-ETP are combined using TSEBRA.
Alignment of RNA-Seq reads
GeneMark-ETP utilizes Stringtie2 to assemble RNA-Seq data, which requires that the aligned reads (BAM files) contain the XS (strand) tag for spliced reads. Therefore, if you align your reads with HISAT2, you must enable the --dta option, or if you use STAR, you must use the --outSAMstrandField intronMotif option. TopHat alignments include this tag by default.
To call the pipeline in this mode, you have to provide it with a protein database using --prot_seq (as described in BRAKER with protein data), and RNA-Seq data either by their SRA ID so that they are downloaded by BRAKER, as unaligned reads in FASTQ format, and/or as aligned reads in bam format (as described in BRAKER with RNA-Seq data). You could also specify already processed extrinsic evidence using the --hints option. However, this is not recommend for a normal BRAKER run in ETPmode, as these hints won't be used in the GeneMark-ETP step. Only use --hints when you want to skip the GenMark-ETP step!
Examples of how you could run BRAKER in ETPmode:
braker.pl --genome=genome.fa --prot_seq=orthodb.fa
--rnaseq_sets_ids=SRA_ID1,SRA_ID2
--rnaseq_sets_dirs=/path/to/local/RNA-Seq/files/
braker.pl --genome=genome.fa --prot_seq=orthodb.fa
--rnaseq_sets_ids=SRA_ID1,SRA_ID2,SRA_ID3
braker.pl --genome=genome.fa --prot_seq=orthodb.fa
--bam=/path/to/SRA_ID1.bam,/path/to/SRA_ID2.bam
A preliminary protocol for integration of assembled subreads from PacBio ccs sequencing in combination with short read Illumina RNA-Seq and protein database is described at https://github.com/Gaius-Augustus/BRAKER/blob/master/docs/long_reads/long_read_protocol.md
We forked GeneMark-ETP and hard coded that StringTie will perform long read assembly in that particular version. If you want to use this 'fast-hack' version for BRAKER, you have to prepare the BAM file with long read to genome spliced alignments outside of BRAKER, eg:
T=48 # adapt to your number of threads
minimap2 -t${T} -ax splice:hq -uf genome.fa isoseq.fa > isoseq.sam
samtools view -bS --threads ${T} isoseq.sam -o isoseq.bam
Pull the adapted container:
singularity build braker3_lr.sif docker://teambraker/braker3:isoseq
Calling BRAKER3 with a BAM file of spliced-aligned IsoSeq Reads:
singularity exec -B ${PWD}:${PWD} braker3_lr.sif braker.pl --genome=genome.fa --prot_seq=protein_db.fa –-bam=isoseq.bam --threads=${T}
Warning Do NOT mix short read and long read data in this BRAKER/GeneMark-ETP variant!
Warning The accuracy of gene prediction here heavily depends on the depth of your isoseq data. We verified with PacBio HiFi reads from 2022 that given sufficient completeness of the assembled transcriptome you will reach similar results as with short reads. However, we also observed a drop in accuracy compared to short reads when using other long read data sets with higher error rates and less sequencing depth.
Please run braker.pl --help to obtain a full list of options.
Compute AUGUSTUS ab initio predictions in addition to AUGUSTUS predictions with hints (additional output files: augustus.ab_initio.* . This may be useful for estimating the quality of training gene parameters when inspecting predictions in a Browser.
One or several command line arguments to be passed to AUGUSTUS, if several arguments are given, separate them by whitespace, ie "--first_arg=sth --second_arg=sth" . This may be be useful if you know that gene prediction in your particular species benefits from a particular AUGUSTUS argument during the prediction step.
Specifies the maximum number of threads that can be used during computation. BRAKER has to run some steps on a single thread, others can take advantage of multiple threads. If you use more than 8 threads, this will not speed up all parallelized steps, in particular, the time consuming optimize_augustus.pl will not use more than 8 threads. However, if you don't mind some threads being idle, using more than 8 threads will speed up other steps.
GeneMark-ETP option: run algorithm with branch point model. Use this option if you genome is a fungus.
Use the present config and parameter files if they exist for 'species'; will overwrite original parameters if BRAKER performs an AUGUSTUS training.
Execute CRF training for AUGUSTUS; resulting parameters are only kept for final predictions if they show higher accuracy than HMM parameters. This increases runtime!
Change the parameter
Generate UTR training examples for AUGUSTUS from RNA-Seq coverage information, train AUGUSTUS UTR parameters and predict genes with AUGUSTUS and UTRs, including coverage information for RNA-Seq as evidence. This is an experimental feature!
If you performed a BRAKER run without --UTR=on, you can add UTR parameter training and gene prediction with UTR parameters (and only RNA-Seq hints) with the following command:
braker.pl --genome=../genome.fa --addUTR=on
--bam=../RNAseq.bam --workingdir=$wd
--AUGUSTUS_hints_preds=augustus.hints.gtf
--threads=8 --skipAllTraining --species=somespecies
Modify augustus.hints.gtf to point to the AUGUSTUS predictions with hints from previous BRAKER run; modify flaning_DNA value to the flanking region from the log file of your previous BRAKER run; modify some_new_working_directory to the location where BRAKER should store results of the additional BRAKER run; modify somespecies to the species name used in your previous BRAKER run.
Add UTRs from RNA-Seq converage information to AUGUSTUS gene predictions using GUSHR. No training of UTR parameters and no gene prediction with UTR parameters is performed.
If you performed a BRAKER run without --addUTR=on, you can add UTRs results of a previous BRAKER run with the following command:
braker.pl --genome=../genome.fa --addUTR=on
--bam=../RNAseq.bam --workingdir=$wd
--AUGUSTUS_hints_preds=augustus.hints.gtf --threads=8
--skipAllTraining --species=somespecies
Modify augustus.hints.gtf to point to the AUGUSTUS predictions with hints from previous BRAKER run; modify some_new_working_directory to the location where BRAKER should store results of the additional BRAKER run; this run will not modify AUGUSTUS parameters. We recommend that you specify the original species of the original run with --species=somespecies . Otherwise, BRAKER will create an unneeded species parameters directory Sp_* .
If --UTR=on is enabled, strand-separated bam-files can be provided with --bam=plus.bam,minus.bam . In that case, --stranded=... should hold the strands of the bam files ( + for plus strand, - for minus strand, . for unstranded). Note that unstranded data will be used in the gene prediction step, only, if the parameter --stranded=... is set. This is an experimental feature! GUSHR currently does not take advantage of stranded data.
If --makehub and [email protected] (with your valid e-mail adress) are provided, a track data hub for visualizing results with the UCSC Genome Browser will be generated using MakeHub (https://github.com/Gaius-Augustus/MakeHub).
By default, GeneMark-ES/ET/EP/ETP uses a probability of 0.001 for predicting the donor splice site pattern GC (instead of GT). It may make sense to increase this value for species where this donor splice site is more common. For example, in the species Emiliania huxleyi , about 50% of donor splice sites have the pattern GC (https://media.nature.com/original/nature-assets/nature/journal/v499/n7457/extref/nature12221-s2.pdf, page 5).
Use a species-specific lineage, eg arthropoda_odb10 for an arthropod. BRAKER does not support auto-typing of the lineage.
Specifying a BUSCO-lineage invokes two changes in BRAKER R28 :
BRAKER will run compleasm with the specified lineage in genome mode and convert the detected BUSCO matches into hints for AUGUSTUS. This may increase the number of BUSCOs in the augustus.hints.gtf file slightly.
BRAKER will invoke best_by_compleasm.py to check whether the braker.gtf file that is by default generated by TSEBRA has the lowest amount of missing BUSCOs compared to the augustus.hints.gtf and the genemark.gtf file. If not, the following decision schema is applied to re-run TSEBRA to minimize the missing BUSCOs in the final output of BRAKER (always braker.gtf). If an alternative and better gene set is created, the original braker.gtf gene set is moved to a directory called braker_original. Information on what happened during the best_by_compleasm.py run is written to the file best_by_compleasm.log.
![best_by_busco[fig14]](https://images.downcodes.com/uploads/20250214/img_67aee79a11fd439.png)
Please note that using BUSCO to assess the quality of a gene set, in particular when comparing BRAKER to other pipelines, does not make sense once you specified a BUSCO lineage. We recommend that you use other measures to assess the quality of your gene set, eg by comparing it to a reference gene set or running OMArk.
BRAKER produces several important output files in the working directory.
braker.gtf: Final gene set of BRAKER. This file may contain different contents depending on how you called BRAKER
in ETPmode: Final gene set of BRAKER consisting of genes predicted by AUGUSTUS and GeneMark-ETP that were combined and filtered by TSEBRA.
otherwise: Union of augustus.hints.gtf and reliable GeneMark-ES/ET/EP predictions (genes fully supported by external evidence). In --esmode , this is the union of augustus.ab_initio.gtf and all GeneMark-ES genes. Thus, this set is generally more sensitive (more genes correctly predicted) and can be less specific (more false-positive predictions can be present). This output is not necessarily better than augustus.hints.gtf, and it is not recommended to use it if BRAKER was run in ESmode.
braker.codingseq: Final gene set with coding sequences in FASTA format
braker.aa: Final gene set with protein sequences in FASTA format
braker.gff3: Final gene set in gff3 format (only produced if the flag --gff3 was specified to BRAKER.
Augustus/*: Augustus gene set(s) in as gtf/conding/aa files
GeneMark-E*/genemark.gtf: Genes predicted by GeneMark-ES/ET/EP/EP+/ETP in GTF-format.
hintsfile.gff: The extrinsic evidence data extracted from RNAseq.bam and/or protein data.
braker_original/*: Genes predicted by BRAKER (TSEBRA merge) before compleasm was used to improve BUSCO completeness
bbc/*: output folder of best_by_compleasm.py script from TSEBRA that is used to improve BUSCO completeness in the final output of BRAKER
Output files may be present with the following name endings and formats:
Coding sequences in FASTA-format are produced if the flag --skipGetAnnoFromFasta was not set.
Protein sequence files in FASTA-format are produced if the flag --skipGetAnnoFromFasta was not set.
For details about gtf format, see http://www.sanger.ac.uk/Software/formats/GFF/. A GTF-format file contains one line per predicted exon. Exemplo:
HS04636 AUGUSTUS initial 966 1017 . + 0 transcript_id "g1.1"; gene_id "g1";
HS04636 AUGUSTUS internal 1818 1934 . + 2 transcript_id "g1.1"; gene_id "g1";
The columns (fields) contain:
seqname source feature start end score strand frame transcript ID and gene ID
If the --makehub option was used and MakeHub is available on your system, a hub directory beginning with the name hub_ will be created. Copy this directory to a publicly accessible web server. A file hub.txt resides in the directory. Provide the link to that file to the UCSC Genome Browser for visualizing results.
An incomplete example data set is contained in the directory BRAKER/example . In order to complete the data set, please download the RNA-Seq alignment file (134 MB) with wget :
cd BRAKER/example
wget http://topaz.gatech.edu/GeneMark/Braker/RNAseq.bam
In case you have trouble accessing that file, there's also a copy available from another server:
cd BRAKER/example
wget http://bioinf.uni-greifswald.de/augustus/datasets/RNAseq.bam
The example data set was not compiled in order to achieve optimal prediction accuracy, but in order to quickly test pipeline components. The small subset of the genome used in these test examples is not long enough for BRAKER training to work well.
Data corresponds to the last 1,000,000 nucleotides of Arabidopsis thaliana 's chromosome Chr5, split into 8 artificial contigs.
RNA-Seq alignments were obtained by VARUS.
The protein sequences are a subset of OrthoDB v10 plants proteins.
List of files:
genome.fa - genome file in fasta formatRNAseq.bam - RNA-Seq alignment file in bam format (this file is not a part of this repository, it must be downloaded separately from http://topaz.gatech.edu/GeneMark/Braker/RNAseq.bam)RNAseq.hints - RNA-Seq hints (can be used instead of RNAseq.bam as RNA-Seq input to BRAKER)proteins.fa - protein sequences in fasta formatThe below given commands assume that you configured all paths to tools by exporting bash variables or that you have the necessary tools in your $PATH.
The example data set also contains scripts tests/test*.sh that will execute below listed commands for testing BRAKER with the example data set. You find example results of AUGUSTUS and GeneMark-ES/ET/EP/ETP in the folder results/test* . Be aware that BRAKER contains several parts where random variables are used, ie results that you obtain when running the tests may not be exactly identical. To compare your test results with the reference ones, you can use the compare_intervals_exact.pl script as follows:
# Compare CDS features
compare_intervals_exact.pl --f1 augustus.hints.gtf --f2 ../../results/test${N}/augustus.hints.gtf --verbose
# Compare transcripts
compare_intervals_exact.pl --f1 augustus.hints.gtf --f2 ../../results/test${N}/augustus.hints.gtf --trans --verbose
Several tests use --gm_max_intergenic 10000 option to make the test runs faster. It is not recommended to use this option in real BRAKER runs, the speed increase achieved by adjusting this option is negligible on full-sized genomes.
We give runtime estimations derived from computing on Intel(R) Xeon(R) CPU E5530 @ 2.40GHz .
The following command will run the pipeline according to Figure 3:
braker.pl --genome genome.fa --bam RNAseq.bam --threads N --busco_lineage=lineage_odb10
This test is implemented in test1.sh , expected runtime is ~20 minutes.
The following command will run the pipeline according to Figure 4:
braker.pl --genome genome.fa --prot_seq proteins.fa --threads N --busco_lineage=lineage_odb10
This test is implemented in test2.sh , expected runtime is ~20 minutes.
The following command will run a pipeline that first trains GeneMark-ETP with protein and RNA-Seq hints and subsequently trains AUGUSTUS on the basis of GeneMark-ETP predictions. AUGUSTUS predictions are also performed with hints from both sources, see Figure 5.
Run with local RNA-Seq file:
braker.pl --genome genome.fa --prot_seq proteins.fa --bam ../RNAseq.bam --threads N --busco_lineage=lineage_odb10
This test is implemented in test3.sh , expected runtime is ~20 minutes.
Download RNA-Seq library from Sequence Read Archive (~1gb):
braker.pl --genome genome.fa --prot_seq proteins.fa --rnaseq_sets_ids ERR5767212 --threads N --busco_lineage=lineage_odb10
This test is implemented in test3_4.sh , expected runtime is ~35 minutes.
The training step of all pipelines can be skipped with the option --skipAllTraining . This means, only AUGUSTUS predictions will be performed, using pre-trained, already existing parameters. For example, you can predict genes with the command:
braker.pl --genome=genome.fa --bam RNAseq.bam --species=arabidopsis
--skipAllTraining --threads N
This test is implemented in test4.sh , expected runtime is ~1 minute.
The following command will run the pipeline with no extrinsic evidence:
braker.pl --genome=genome.fa --esmode --threads N
This test is implemented in test5.sh , expected runtime is ~20 minutes.
The following command will run BRAKER with training UTR parameters from RNA-Seq coverage data:
braker.pl --genome genome.fa --bam RNAseq.bam --UTR=on --threads N
This test is implemented in test6.sh , expected runtime is ~20 minutes.
The following command will add UTRs to augustus.hints.gtf from RNA-Seq coverage data:
braker.pl --genome genome.fa --bam RNAseq.bam --addUTR=on --threads N
This test is implemented in test7.sh , expected runtime is ~20 minutes.
There is currently no clean way to restart a failed BRAKER run (after solving some problem). However, it is possible to start a new BRAKER run based on results from a previous run -- given that the old run produced the required intermediate results. We will in the following refer to the old working directory with variable ${BRAKER_OLD} , and to the new BRAKER working directory with ${BRAKER_NEW} . The file what-to-cite.txt will always only refer to the software that was actually called by a particular run. You might have to combine the contents of ${BRAKER_NEW}/what-to-cite.txt with ${BRAKER_OLD}/what-to-cite.txt for preparing a publication. The following figure illustrates at which points BRAKER run may be intercepted.
![braker-intercept[fig8]](https://images.downcodes.com/uploads/20250214/img_67aee79a12cab310.png)
Figure 10: Points for intercepting a BRAKER run and reusing intermediate results in a new BRAKER run.
This option is only possible for BRAKER in ETmode or EPmode and não in ETPmode!
If you have access to an existing BRAKER output that contains hintsfiles that were generated from extrinsic data, such as RNA-Seq or protein sequences, you can recycle these hints files in a new BRAKER run. Also, hints from a separate ProtHint run can be directly used in BRAKER.
The hints can be given to BRAKER with --hints ${BRAKER_OLD}/hintsfile.gff option. This is illustrated in the test files test1_restart1.sh , test2_restart1.sh , test4_restart1.sh . The other modes (for which this test is missing) cannot be restarted in this way.
The GeneMark result can be given to BRAKER with --geneMarkGtf ${BRAKER_OLD}/GeneMark*/genemark.gtf option if BRAKER is run in ETmode or EPmode. This is illustrated in the test files test1_restart2.sh , test2_restart2.sh , test5_restart2.sh .
In ETPmode, you can either provide BRAKER with the results of the GeneMarkETP step manually, with --geneMarkGtf ${BRAKER_OLD}/GeneMark-ETP/proteins.fa/genemark.gtf , --traingenes ${BRAKER_OLD}/GeneMark-ETP/training.gtf , and --hints ${BRAKER_OLD}/hintsfile.gff (see test3_restart1.sh for an example), or you can specify the previous GeneMark-ETP results with the option --gmetp_results_dir ${BRAKER_OLD}/GeneMark-ETP/ so that BRAKER can search for the files automatically (see test3_restart2.sh for an example).
The trained species parameters for AGUSTUS can be passed with --skipAllTraining and --species $speciesName options. This is illustrated in test*_restart3.sh files. Note that in ETPmode you have to specify the GeneMark files as described in Option 2!
Before reporting bugs, please check that you are using the most recent versions of GeneMark-ES/ET/EP/ETP, AUGUSTUS and BRAKER. Also, check the list of Common problems, and the Issue list on GitHub before reporting bugs. We do monitor open issues on GitHub. Sometimes, we are unable to help you, immediately, but we try hard to solve your problems.
If you found a bug, please open an issue at https://github.com/Gaius-Augustus/BRAKER/issues (or contact [email protected] or [email protected]).
Information worth mentioning in your bug report:
Check in braker/yourSpecies/braker.log at which step braker.pl crashed.
There are a number of other files that might be of interest, depending on where in the pipeline the problem occurred. Some of the following files will not be present if they did not contain any errors.
braker/yourSpecies/errors/bam2hints.*.stderr - will give details on a bam2hints crash (step for converting bam file to intron gff file)
braker/yourSpecies/hintsfile.gff - is this file empty? If yes, something went wrong during hints generation - does this file contain hints from source “b2h” and of type “intron”? If not: GeneMark-ET will not be able to execute properly. Conversely, GeneMark-EP+ will not be able to execute correctly if hints from the source "ProtHint" are missing.
braker/yourSpecies/spaln/*err - errors reported by spaln
braker/yourSpecies/errors/GeneMark-{ET,EP,ETP}.stderr - errors reported by GeneMark-ET/EP+/ETP
braker/yourSpecies/errors/GeneMark-{ET,EP,ETP).stdout - may give clues about the point at which errors in GeneMark-ET/EP+/ETP occured
braker/yourSpecies/GeneMark-{ET,EP,ETP}/genemark.gtf - is this file empty? If yes, something went wrong during executing GeneMark-ET/EP+/ETP
braker/yourSpecies/GeneMark-{ET,EP}/genemark.f.good.gtf - is this file empty? If yes, something went wrong during filtering GeneMark-ET/EP+ genes for training AUGUSTUS
braker/yourSpecies/genbank.good.gb - try a “grep -c LOCUS genbank.good.gb” to determine the number of training genes for training AUGUSTUS, should not be low
braker/yourSpecies/errors/firstetraining.stderr - contains errors from first iteration of training AUGUSTUS
braker/yourSpecies/errors/secondetraining.stderr - contains errors from second iteration of training AUGUSTUS
braker/yourSpecies/errors/optimize_augustus.stderr - contains errors optimize_augustus.pl (additional training set for AUGUSTUS)
braker/yourSpecies/errors/augustus*.stderr - contain AUGUSTUS execution errors
braker/yourSpecies/startAlign.stderr - if you provided a protein fasta file, something went wrong during protein alignment
braker/yourSpecies/startAlign.stdout - may give clues on at which point protein alignment went wrong
BRAKER complains that the RNA-Seq file does not correspond to the provided genome file, but I am sure the files correspond to each other!
Please check the headers of the genome FASTA file. If the headers are long and contain whitespaces, some RNA-Seq alignment tools will truncate sequence names in the BAM file. This leads to an error with BRAKER. Solution: shorten/simplify FASTA headers in the genome file before running the RNA-Seq alignment and BRAKER.
GeneMark fails!
(a) GeneMark by default only uses contigs longer than 50k for training. If you have a highly fragmented assembly, this might lead to "no data" for training. You can override the default minimal length by setting the BRAKER argument --min_contig=10000 .
(b) see "[something] failed to execute" below.
[something] failed to execute!
When providing paths to software to BRAKER, please use absolute, non-abbreviated paths. For example, BRAKER might have problems with --SAMTOOLS_PATH=./samtools/ or --SAMTOOLS_PATH=~/samtools/ . Please use SAMTOOLS_PATH=/full/absolute/path/to/samtools/ , instead. This applies to all path specifications as command line options to braker.pl . Relative paths and absolute paths will not pose problems if you export a bash variable, instead, or if you append the location of tools to your $PATH variable.
GeneMark-ETP in BRAKER dies with '/scratch/11232323': No such file or directory.
This appears to be related to sorting large files, and it's a system configuration depending problem. Solve it with export TMPDIR=/tmp/ before calling BRAKER via Singularity.
BRAKER cannot find the Augustus script XYZ...
Update Augustus from github with git clone https://github.com/Gaius-Augustus/Augustus.git . Do not use Augustus from other sources. BRAKER is highly dependent on an up-to-date Augustus. Augustus releases happen rather rarely, updates to the Augustus scripts folder occur rather frequently.
Does BRAKER depend on Python3?
Isso faz. The python scripts employed by BRAKER are not compatible with Python2.
Why does BRAKER predict more genes than I expected?
If transposable elements (or similar) have not been masked appropriately, AUGUSTUS tends to predict those elements as protein coding genes. This can lead to a huge number genes. You can check whether this is the case for your project by BLASTing (or DIAMONDing) the predicted protein sequences against themselves (all vs. all) and counting how many of the proteins have a high number of high quality matches. You can use the output of this analysis to divide your gene set into two groups: the protein coding genes that you want to find and the repetitive elements that were additionally predicted.
I am running BRAKER in Anaconda and something fails...
Update AUGUSTUS and BRAKER from github with git clone https://github.com/Gaius-Augustus/Augustus.git and git clone https://github.com/Gaius-Augustus/BRAKER.git . The Anaconda installation is great, but it relies on releases of AUGUSTUS and BRAKER - which are often lagging behind. Please use the current GitHub code, instead.
Why and where is the GenomeThreader support gone?
BRAKER is a joint project between teams from University of Greifswald and Georgia Tech. While the group of Mark Bordovsky from Georgia Tech contributes GeneMark expertise, the group of Mario Stanke from University of Greifswald contributes AUGUSTUS expertise. Using GenomeThreader to build training genes for AUGUSTUS in BRAKER circumvents execution of GeneMark. Thus, the GenomeThreader mode is strictly speaking not part of the BRAKER project. The previous functionality of BRAKER with GenomeThreader has been moved to GALBA at https://github.com/Gaius-Augustus/GALBA. Note that GALBA has also undergone extension for using Miniprot instead of GenomeThreader.
My BRAKER gene set has too many BUSCO duplicates!
AUGUSTUS within BRAKER can predict alternative splicing isoforms. Also the merge of the AUGUSTUS and GeneMark gene set by TSEBRA within BRAKER may result in additional isoforms for a single gene. The BUSCO duplicates usually come from alternative splicing isoforms, ie they are expected.
Augustus and/or etraining within BRAKER complain that the file aug_cmdln_parameters.json is missing. Even though I am using the latest Singularity container!
BRAKER copies the AUGUSTUS_CONFIG_PATH folder to a writable location. In older versions of Augustus, that file was indeed not existing. If the local writable copy of a folder already exists, BRAKER will not re-copy it. Simply delete the old folder. (It is often ~/.augustus , so you can simply do rm -rf ~/.augustus ; the folder might be residing in $PWD if your home directory was not writable).
I sit behind a firewall, compleasm cannot download the BUSCO files, what can I do? See Issue #785 (comment)
Since BRAKER is a pipeline that calls several Bioinformatics tools, publication of results obtained by BRAKER requires that not only BRAKER is cited, but also the tools that are called by BRAKER. BRAKER will output a file what-to-cite.txt in the BRAKER working directory, informing you about which exact sources apply to your run.
Always cite:
Stanke, M., Diekhans, M., Baertsch, R. and Haussler, D. (2008). Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics, doi: 10.1093/bioinformatics/btn013.
Stanke. M., Schöffmann, O., Morgenstern, B. and Waack, S. (2006). Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics 7, 62.
If you provided any kind of evidence for BRAKER, cite:
If you provided both short read RNA-Seq evidence and a large database of proteins, cite:
Gabriel, L., Bruna, T., Hoff, KJ, Ebel, M., Lomsadze, A., Borodovsky, M., Stanke, M. (2023). BRAKER3: Fully Automated Genome Annotation Using RNA-Seq and Protein Evidence with GeneMark-ETP, AUGUSTUS and TSEBRA. bioRxiV, doi: 10.1101/2023.06.10.54444910.1101/2023.01.01.474747.
Bruna, T., Lomsadze, A., Borodovsky, M. (2023). GeneMark-ETP: Automatic Gene Finding in Eukaryotic Genomes in Consistence with Extrinsic Data. bioRxiv, doi: 10.1101/2023.01.13.524024.
Kovaka, S., Zimin, AV, Pertea, GM, Razaghi, R., Salzberg, SL, & Pertea, M. (2019). Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome biology, 20(1):1-13.
Pertea, G., & Pertea, M. (2020). GFF utilities: GffRead and GffCompare. F1000Research, 9.
Quinlan, AR (2014). BEDTools: the Swiss‐army tool for genome feature analysis. Current protocols in bioinformatics, 47(1):11-12.
If the only source of evidence for BRAKER was a large database of protein sequences, cite:
If the only source of evidence for BRAKER was RNA-Seq data, cite:
Hoff, KJ, Lange, S., Lomsadze, A., Borodovsky, M. and Stanke, M. (2016). BRAKER1: unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics, 32(5):767-769.
Lomsadze, A., Paul DB, and Mark B. (2014) Integration of Mapped Rna-Seq Reads into Automatic Training of Eukaryotic Gene Finding Algorithm. Nucleic Acids Research 42(15): e119--e119
If you called BRAKER3 with an IsoSeq BAM file, or if you envoked the --busco_lineage option, cite:
If you called BRAKER with the --busco_lineage option, in addition, cite:
Simão, FA, Waterhouse, RM, Ioannidis, P., Kriventseva, EV, & Zdobnov, EM (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics, 31(19), 3210-3212.
Li, H. (2023). Protein-to-genome alignment with miniprot. Bioinformatics, 39(1), btad014.
Huang, N., & Li, H. (2023). compleasm: a faster and more accurate reimplementation of BUSCO. Bioinformatics, 39(10), btad595.
If any kind of AUGUSTUS training was performed by BRAKER, check carefully whether you configured BRAKER to use NCBI BLAST or DIAMOND. One of them was used to filter out redundant training gene structures.
If you used NCBI BLAST, please cite:
Altschul, AF, Gish, W., Miller, W., Myers, EW and Lipman, DJ (1990). A basic local alignment search tool. J Mol Biol 215:403--410.
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., and Madden, TL (2009). Blast+: architecture and applications. BMC bioinformatics, 10(1):421.
If you used DIAMOND, please cite:
If BRAKER was executed with a genome file and no extrinsic evidence, cite, then GeneMark-ES was used, cite:
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, YO and Borodovsky, M. (2005). Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Research, 33(20):6494--6506.
Ter-Hovhannisyan, V., Lomsadze, A., Chernoff, YO and Borodovsky, M. (2008). Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome research, pages gr--081612, 2008.
Hoff, KJ, Lomsadze, A., Borodovsky, M. and Stanke, M. (2019). Whole-Genome Annotation with BRAKER. Methods Mol Biol. 1962:65-95, doi: 10.1007/978-1-4939-9173-0_5.
If BRAKER was run with proteins as source of evidence, please cite all tools that are used by the ProtHint pipeline to generate hints:
Bruna, T., Lomsadze, A., & Borodovsky, M. (2020). GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genomics and Bioinformatics, 2(2), lqaa026.
Buchfink, B., Xie, C., Huson, DH (2015). Fast and sensitive protein alignment using DIAMOND. Nature Methods 12:59-60.
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, YO and Borodovsky, M. (2005). Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Research, 33(20):6494--6506.
Iwata, H., and Gotoh, O. (2012). Benchmarking spliced alignment programs including Spaln2, an extended version of Spaln that incorporates additional species-specific features. Nucleic acids research, 40(20), e161-e161.
Gotoh, O., Morita, M., Nelson, DR (2014). Assessment and refinement of eukaryotic gene structure prediction with gene-structure-aware multiple protein sequence alignment. BMC bioinformatics, 15(1), 189.
If BRAKER was executed with RNA-Seq alignments in bam-format, then SAMtools was used, cite:
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R.; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25(16):2078-9.
Barnett, DW, Garrison, EK, Quinlan, AR, Strömberg, MP and Marth GT (2011). BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics, 27(12):1691-2
If BRAKER downloaded RNA-Seq libraries from SRA using their IDs, cite SRA, SRA toolkit, and HISAT2:
Leinonen, R., Sugawara, H., Shumway, M., & International Nucleotide Sequence Database Collaboration. (2010). The sequence read archive. Nucleic acids research, 39(suppl_1), D19-D21.
SRA Toolkit Development Team (2020). SRA Toolkit. https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software.
Kim, D., Paggi, JM, Park, C., Bennett, C., & Salzberg, SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature biotechnology, 37(8):907-915.
If BRAKER was executed using RNA-Seq data in FASTQ format, cite HISAT2:
If BRAKER called MakeHub for creating a track data hub for visualization of BRAKER results with the UCSC Genome Browser, cite:
If BRAKER called GUSHR for generating UTRs, cite:
Keilwagen, J., Hartung, F., Grau, J. (2019) GeMoMa: Homology-based gene prediction utilizing intron position conservation and RNA-seq data. Methods Mol Biol. 1962:161-177, doi: 10.1007/978-1-4939-9173-0_9.
Keilwagen, J., Wenk, M., Erickson, JL, Schattat, MH, Grau, J., Hartung F. (2016) Using intron position conservation for homology-based gene prediction. Nucleic Acids Research, 44(9):e89.
Keilwagen, J., Hartung, F., Paulini, M., Twardziok, SO, Grau, J. (2018) Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinformatics, 19(1):189.
All source code, ie scripts/*.pl or scripts/*.py are under the Artistic License (see http://www.opensource.org/licenses/artistic-license.php).
[F1] EX = ES/ET/EP/ETP, all available for download under the name GeneMark-ES/ET/EP ↩
[F2] Please use the latest version from the master branch of AUGUSTUS distributed by the original developers, it is available from github at https://github.com/Gaius-Augustus/Augustus. Problems have been reported from users that tried to run BRAKER with AUGUSTUS releases maintained by third parties, ie Bioconda. ↩
[F4] install with sudo apt-get install cpanminus ↩
[F6] The binary may eg reside in bamtools/build/src/toolkit ↩
[R0] Bruna, Tomas, Hoff, Katharina J., Lomsadze, Alexandre, Stanke, Mario, and Borodovsky, Mark. 2021. “BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database." NAR Genomics and Bioinformatics 3(1):lqaa108.↩
[R1] Hoff, Katharina J, Simone Lange, Alexandre Lomsadze, Mark Borodovsky, and Mario Stanke. 2015. “BRAKER1: Unsupervised Rna-Seq-Based Genome Annotation with Genemark-et and Augustus.” Bioinformatics 32 (5). Oxford University Press: 767--69.↩
[R2] Lomsadze, Alexandre, Paul D Burns, and Mark Borodovsky. 2014. “Integration of Mapped Rna-Seq Reads into Automatic Training of Eukaryotic Gene Finding Algorithm.” Nucleic Acids Research 42 (15). Oxford University Press: e119--e119.↩
[R3] Stanke, Mario, Mark Diekhans, Robert Baertsch, and David Haussler. 2008. “Using Native and Syntenically Mapped cDNA Alignments to Improve de Novo Gene Finding.” Bioinformatics 24 (5). Oxford University Press: 637--44.↩
[R4] Stanke, Mario, Oliver Schöffmann, Burkhard Morgenstern, and Stephan Waack. 2006. “Gene Prediction in Eukaryotes with a Generalized Hidden Markov Model That Uses Hints from External Sources.” BMC Bioinformatics 7 (1). BioMed Central: 62.↩
[R5] Barnett, Derek W, Erik K Garrison, Aaron R Quinlan, Michael P Strömberg, and Gabor T Marth. 2011. “BamTools: A C++ Api and Toolkit for Analyzing and Managing Bam Files.” Bioinformatics 27 (12). Oxford University Press: 1691--2.↩
[R6] Li, Heng, Handsaker, Bob, Alec Wysoker, Tim Fennell, Jue Ruan, Nils Homer, Gabor Marth, Goncalo Abecasis, and Richard Durbin. 2009. “The Sequence Alignment/Map Format and Samtools.” Bioinformatics 25 (16). Oxford University Press: 2078--9.↩
[R7] Gremme, G. 2013. “Computational Gene Structure Prediction.” PhD thesis, Universität Hamburg.↩
[R8] Gotoh, Osamu. 2008a. “A Space-Efficient and Accurate Method for Mapping and Aligning cDNA Sequences onto Genomic Sequence.” Nucleic Acids Research 36 (8). Oxford University Press: 2630--8.↩
[R9] Iwata, Hiroaki, and Osamu Gotoh. 2012. “Benchmarking Spliced Alignment Programs Including Spaln2, an Extended Version of Spaln That Incorporates Additional Species-Specific Features.” Nucleic Acids Research 40 (20). Oxford University Press: e161--e161.↩
[R10] Osamu Gotoh. 2008b. “Direct Mapping and Alignment of Protein Sequences onto Genomic Sequence.” Bioinformatics 24 (21). Oxford University Press: 2438--44.↩
[R11] Slater, Guy St C, and Ewan Birney. 2005. “Automated Generation of Heuristics for Biological Sequence Comparison.” BMC Bioinformatics 6(1). BioMed Central: 31.↩
[R12] Altschul, SF, W. Gish, W. Miller, EW Myers, and DJ Lipman. 1990. “Basic Local Alignment Search Tool.” Journal of Molecular Biology 215:403--10.↩
[R13] Camacho, Christiam, et al. 2009. “BLAST+: architecture and applications.“ BMC Bioinformatics 1(1): 421.↩
[R14] Lomsadze, A., V. Ter-Hovhannisyan, YO Chernoff, and M. Borodovsky. 2005. “Gene identification in novel eukaryotic genomes by self-training algorithm.” Nucleic Acids Research 33 (20): 6494--6506. doi:10.1093/nar/gki937.↩
[R15] Ter-Hovhannisyan, Vardges, Alexandre Lomsadze, Yury O Chernoff, and Mark Borodovsky. 2008. “Gene Prediction in Novel Fungal Genomes Using an Ab Initio Algorithm with Unsupervised Training.” Genome Research . Cold Spring Harbor Lab, gr--081612.↩
[R16] Hoff, KJ 2019. MakeHub: Fully automated generation of UCSC Genome Browser Assembly Hubs. Genomics, Proteomics and Bioinformatics , in press, preprint on bioarXive, doi: https://doi.org/10.1101/550145.↩
[R17] Bruna, T., Lomsadze, A., & Borodovsky, M. 2020. GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genomics and Bioinformatics, 2(2), lqaa026. doi: https://doi.org/10.1093/nargab/lqaa026.↩
[R18] Kriventseva, EV, Kuznetsov, D., Tegenfeldt, F., Manni, M., Dias, R., Simão, FA, and Zdobnov, EM 2019. OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Research, 47(D1), D807-D811.↩
[R19] Keilwagen, J., Hartung, F., Grau, J. (2019) GeMoMa: Homology-based gene prediction utilizing intron position conservation and RNA-seq data. Methods Mol Biol. 1962:161-177, doi: 10.1007/978-1-4939-9173-0_9.↩
[R20] Keilwagen, J., Wenk, M., Erickson, JL, Schattat, MH, Grau, J., Hartung F. (2016) Using intron position conservation for homology-based gene prediction. Nucleic Acids Research, 44(9):e89.↩
[R21] Keilwagen, J., Hartung, F., Paulini, M., Twardziok, SO, Grau, J. (2018) Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinformatics, 19(1):189.↩
[R22] SRA Toolkit Development Team (2020). SRA Toolkit. https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software.[↩](#a22)
[R23] Kim, D., Paggi, JM, Park, C., Bennett, C., & Salzberg, SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature biotechnology, 37(8):907-915.↩
[R24] Quinlan, AR (2014). BEDTools: the Swiss‐army tool for genome feature analysis. Current protocols in bioinformatics, 47(1):11-12.↩
[R25] Kovaka, S., Zimin, AV, Pertea, GM, Razaghi, R., Salzberg, SL, & Pertea, M. (2019). Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome biology, 20(1):1-13.↩
[R26] Pertea, G., & Pertea, M. (2020). GFF utilities: GffRead and GffCompare. F1000Research, 9.↩
[R27] Huang, N., & Li, H. (2023). compleasm: a faster and more accurate reimplementation of BUSCO. Bioinformatics, 39(10), btad595.↩
[R28] Bruna, T., Gabriel, L. & Hoff, KJ (2024). Navigating Eukaryotic Genome Annotation Pipelines: A Route Map to BRAKER, Galba, and TSEBRA. arXiv, https://doi.org/10.48550/arXiv.2403.19416 .↩


