binnacle
1.0.0
Para
O binnacle calcula com precisão a cobertura de andaimes de gráficos e se integra perfeitamente aos métodos de liderança de binning, como metabat2, maxbin 2.0 e inventário. O uso de andaimes de gráficos, em oposição aos contigs (abordagem mais comum) para binning, melhora a contiguidade e a qualidade das caixas metagenômicas e pode capturar um conjunto mais amplo dos elementos acessórios dos genomas reconstruídos.
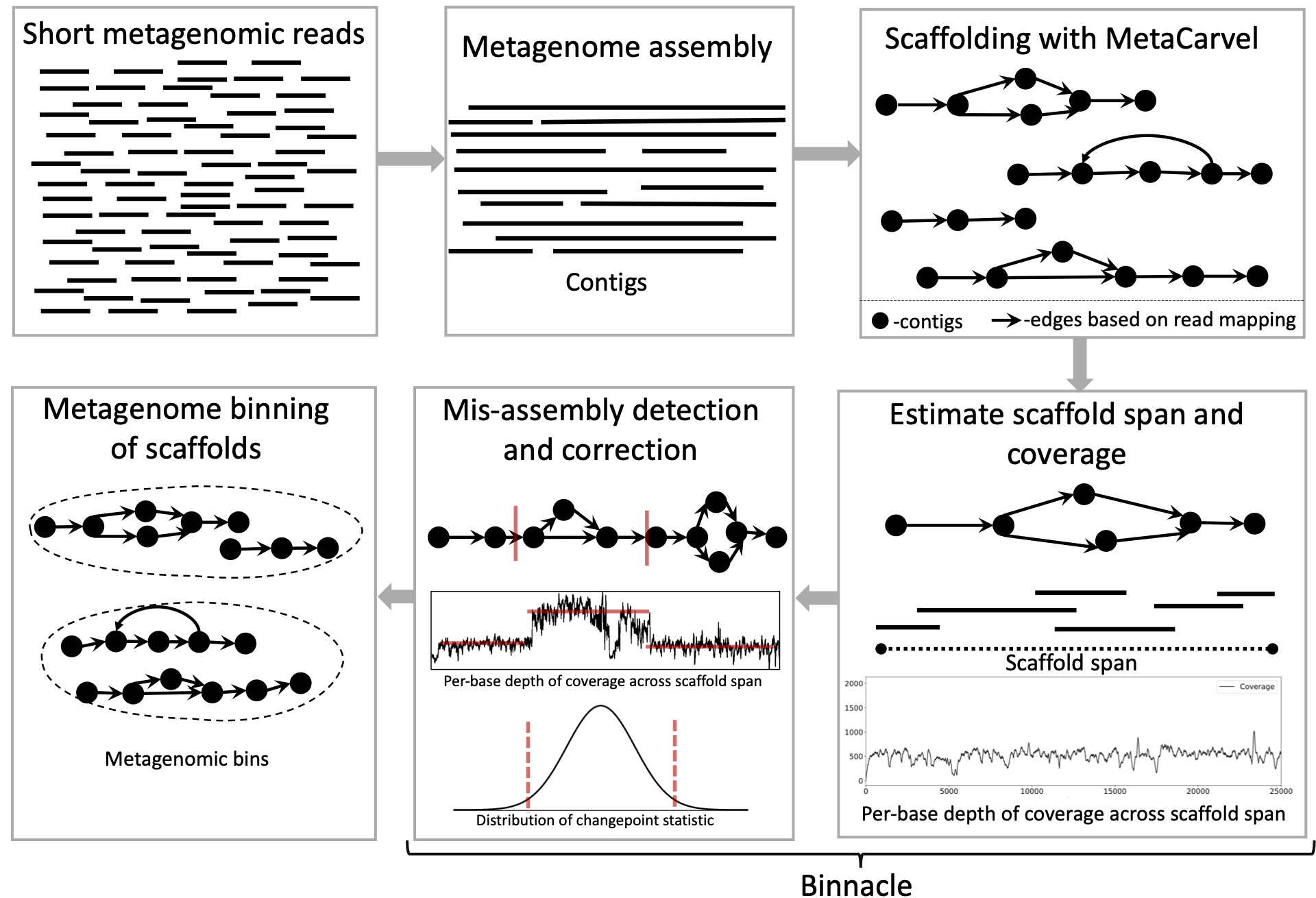
Para executar o Binnacle, você precisará de Python 3.7.x, Bedtools, Samtools, Biopython, Networkx, Numpy e Pandas.
Um arquivo Environment.yml está disponível e isso pode ser usado para criar um ambiente de conda adequado para executar o Binnacle. A documentação detalhada sobre como instalar esses pacotes é fornecida aqui. Utilizamos andaimes de gráficos que são emitidos da ferramenta de andaime metacarvel, portanto, você também precisará baixar e instalar o Metacarvel. Há um guia de instalação passo a passo para metacarvel.
Geralmente, quando você possui uma ou várias amostras metagenômicas, precisamos montar, andaimes e contigs/andaimes de bin de cada amostra para gerar caixas metagenômicas. Recomendamos o uso de megahit para Assembly e Metacarvel para andaimes. Fornecemos um guia auxiliar para trabalhar através de etapas de estimativa de montagem, andaimes e de estimativa de cobertura por base aqui.
Siga estas etapas para gerar arquivos para a execução de métodos de binning com andaimes gráficos:
python Estimate_Abundances.py -g [ORIENTED.gml] -a [COVERAGE_SORTED.txt] -c [CONTIGS.fa] -d [OUTPUT_DIRECTORY]
usage: Estimate_Abundances.py [-h] [-g ASSEMBLY] [-a COVERAGE] [-bam BAMFILE]
[-bed BEDFILE] [-c CONTIGS] -d DIR [-o COORDS]
[-w WINDOW_SIZE] [-t THRESHOLD]
[-n NEIGHBOR_CUTOFF] [-p POSCUTOFF]
[-pre PREFIX]
binnacle: A tool for binning metagenomic datasets using assembly graphs and
scaffolds generated by metacarvel. Estimate_Abundances.py estimates abundance
for scaffolds generated by MetaCarvel. If the coordinates computed by binnacle
is specified then the abundance for each scaffold is estimated based on the
contig abundances and the coordinates. If the coordinates are not specified
then binnacle etimates the abundance from scratch. While calculating all vs
all abundances please specify the coordinates(Coordinates_After_Delinking.txt)
through the "coords" parameter. The abundances can be provided as a bed file,
bam file or a text file describing the per base coverage obtained by running
the genomeCoverageBed program of the bedtools suite.
optional arguments:
-h, --help show this help message and exit
-g ASSEMBLY, --assembly ASSEMBLY
Assembly Graph generated by Metacarvel
-a COVERAGE, --coverage COVERAGE
Output generated by running genomecov -d on the bed
file generated by MetaCarvel.
-bam BAMFILE, --bamfile BAMFILE
Bam file from aligning reads to contigs
-bed BEDFILE, --bedfile BEDFILE
Bed file from aligning reads to contigs. If bed file
is provided please provide a fasta file of the contigs
-c CONTIGS, --contigs CONTIGS
Contigs generated by the assembler, contigs.fasta
-d DIR, --dir DIR output directory for results
-o COORDS, --coords COORDS
Coordinate file generated by Binnacle
-w WINDOW_SIZE, --window_size WINDOW_SIZE
Size of the sliding window for computing test
statistic to identify changepoints in coverages
(Default=1500)
-t THRESHOLD, --threshold THRESHOLD
Threshold to identify outliers (Default=99)
-n NEIGHBOR_CUTOFF, --neighbor_cutoff NEIGHBOR_CUTOFF
Filter size to identify outliers within (Defualt=100)
-p POSCUTOFF, --poscutoff POSCUTOFF
Position cutoff to consider delinking (Default=100)
-pre PREFIX, --prefix PREFIX
Prefix to be attached to all outputs
-g Path to oriented.gml from running metacarvel on sample
-c Path to contigs obtained by assembling reads of sample
-a Coverage of contigs in ths sample by mapping to its reads -- See Wiki for how to calculate coverage information
-d Output directory
-a Coverage of contigs in Sample 1 by mapping reads of Sample 2 -- See Wiki for how to calculate coverage information
-o Coordinates of scaffolds from Sample 1 that you would have generated from the previous step.
-d Same output directory as Sample 1
python Collate.py -h
usage: Collate.py [-h] -d DIR [-m METHOD] [-k KEEP]
binnacle: A tool for binning metagenomic datasets using assembly graphs and
scaffolds generated by metacarvel.Estimate_Abundances.py estimates abundance
for scaffolds generated by MetaCarvel. The program Collate.py collects the
summary files generated by Estimate_Abundances.py
optional arguments:
-h, --help show this help message and exit
-d DIR, --dir DIR Output directory that contains the summary files
generated by running Estimate_Abundances.py
-m METHOD, --method METHOD
Binning method to format the output to. Presently we
support 1. Metabat 2. Maxbin 3. Concoct 4. Binnacle
(Default)
-k KEEP, --keep KEEP Retain the summary files generated by
Estimate_Abundances.py. Defaults to True
Verifique o wiki para obter uma descrição detalhada na configuração do ambiente Python, métodos para calcular a cobertura e um fluxo de trabalho típico para executar o Binnacle.
Para visualizar os andaimes de gráficos, recomendamos o uso do MetaGenomescope, que é um navegador baseado na Web. A entrada no MetaGenomescope é assembly_graph_filtered.gml. A documentação detalhada sobre a instalação e a execução do MetaGenomescope é fornecida aqui.
Cite Muralidharan HS, Shah N, Meisel JS e Pop M (2021) Binnacle: Usando andaimes para melhorar a contiguidade e a qualidade das caixas metagenômicas. Frente. Microbiol. 12: 638561. doi: 10.3389/fmicb.2021.638561.
Esta ferramenta ainda está em desenvolvimento. Abra a edição aqui no Github ou entre em contato conosco se tiver alguma dúvida.
Harihara muralidharan: [email protected]
Nidhi shah: [email protected]


