ClockstaR
1.0.0

Себастьян Дюшен, Мартина Молак и Саймон Ю.В. Хо.
Лаборатория молекулярной экологии, эволюции и филогенетики (MEEP)
Школа биологических наук
Сиднейский университет
10 июня, 2015 г.
partition_data_partitionfinder('drag fasta file with concatenated data here', 'drag partition finder output here')
optim.trees.interactive(folder.parts = 'path to your folder with fasta files and tree topology here')
Реализуйте оптимизацию расстояний между деревьями, используя производную расстояния BSD.
Реализуйте параллельную версию для топологического расстояния
Написать руководство по топологической дистанционной кластеризации
Интегрируйте генератор моделей для тестирования моделей.
Интегрируйте RaxML для максимальной оптимизации длин ветвей и топологий.
Оценка временных масштабов эволюции с помощью наборов мультигенных данных является обычным занятием в филогенетических исследованиях. Наборы мультигенных данных можно разделить по гену, положению кодона или по тому и другому. В этом руководстве мы называем «подмножествами данных» отдельные гены или любую субъединицу мультигенного набора данных. Термин «разделы» будет относиться к группе подмножеств данных.
Хотя подмножества данных могут быть объединены и проанализированы с помощью одной модели с ослабленными часами, закономерности изменения скорости между линиями могут различаться между подмножествами данных, даже если их древовидные топологии идентичны. Например, вариация скорости митохондриальных генов между линиями может отличаться от вариаций ядерных генов. Таким образом, разные модели расслабленных часов могут быть присвоены разным подмножествам данных, чтобы улучшить оценки эволюционных временных масштабов и статистическое соответствие (см. Duchene and Ho., 2014a).
Существует большое количество способов разделения мультигенных наборов данных. Распространенным подходом к сравнению схем разделения является использование факторов Байеса или критериев, основанных на правдоподобии, для подбора модели. Однако в большинстве случаев невозможно протестировать все возможные схемы разделения, особенно с использованием трудоемких методов расчета факторов Байеса.
ClockstaR оценивает длину филогенетических ветвей каждого подмножества данных. Расстояние между ветвями, известное как sBSDmin, рассчитывается для каждой пары деревьев как мера разницы в их закономерностях изменения скорости между линиями. Эти расстояния используются для определения наилучшей стратегии разделения с использованием статистики GAP с алгоритмом кластеризации PAM, реализованным в кластере пакетов (Maechler et al., 2012) (подробнее о метрике sBSDmin см. Duchene et al., 2014b). .
ClockstaR — это пакет R для филогенетического анализа молекулярных часов наборов мультигенных данных. Он использует закономерности изменения скорости между линиями для разных генов, чтобы выбрать стратегию разделения часов. В методе используется метрика расстояния филогенетического дерева и алгоритм машинного обучения без учителя для определения оптимального количества часовых частей и того, какие гены следует анализировать в каждой из частей. Методика разделения, выбранная в ClocsktaR, может использоваться для последующего анализа молекулярных часов с помощью таких программ, как BEAST, MrBayes, PhyloBayes и других.
Пожалуйста, перейдите по этой ссылке для получения оригинальной публикации.
ClockstaR требует установки R. Для этого также требуются некоторые зависимости R, которые можно получить через R, как описано ниже.
Любые запросы или вопросы направляйте Себастьяну Дюшену (sebastian.duchene[at]sydney.edu.au). Некоторое другое программное обеспечение и ресурсы можно найти в Лаборатории молекулярной экологии, эволюции и филогенетики Сиднейского университета.
Загрузите этот репозиторий в виде zip-файла и разархивируйте его. В следующих инструкциях используется папка clockstar_example_data, которая содержит несколько файлов fasta и филогенетическое дерево в формате Newick. Откройте любой из этих файлов в текстовом редакторе, например в Text Wrangler. Эти данные были смоделированы по четырем закономерностям эволюционного изменения скорости. Обратите внимание, что дерево представляет собой древовидную топологию для всех генов или разделов данных. Чтобы запустить ClockstaR, отформатируйте данные аналогично данным примера в clockstar_example_data.
ClockstaR можно установить прямо с GitHub. Для этого требуется пакет devtools. Введите следующий код в командной строке R, чтобы установить все необходимые инструменты (обратите внимание, что для прямой загрузки пакетов вам потребуется подключение к Интернету):.
install . packages ( " devtools " )
library (devtools)
install_github ( ' ClockstaR ' , ' sebastianduchene ' )После скачивания и установки загрузите ClockstaR с библиотекой функций.
library (ClockstaR2)Чтобы увидеть пример запуска программы, введите:
example (ClockstaR2)В оставшейся части этого руководства используется папка clockstar_example_data.
Первым шагом является получение генных деревьев для каждого из выравниваний. Для этого мы используем топологию дерева и оптимизируем длину ветвей, используя выравнивание каждого отдельного гена, в данном случае от A1.fasta до C3.fasta. Если у вас есть деревья генов, сохраните их в файле в формате Ньюика и переходите к следующему шагу (интерактивный запуск clockstar).
Введите следующий код в командной строке R и нажмите Enter:
optim . trees . interactive ()Если вы получили сообщение об ошибке при установке пакета phangorn, используйте этот код, а затем повторите optim.trees.interactive().
install . packcages ( " phangorn " )ClockstaR напечатает следующее сообщение:
Please drag a folder with the data subsets and a tree topology . The files should be in FASTA format, and the trees in NEWICKПеретащите папку clockstar_example_data в консоль R и введите Enter. Обратите внимание, что папка должна содержать только файлы в формате FASTA и древовидную топологию в формате NEWICK. Вы увидите следующее сообщение:
What should be the name of the file to save the optimised trees ?Введите имя файла оптимизированных деревьев. В этом случае мы будем использовать «example.trees».
example . treesНа этом этапе ClockstaR спросит, следует ли использовать отдельную модель замены для каждого гена или использовать JC во всех случаях. Поскольку эти данные были смоделированы в JC, мы наберем «n» и нажмем Enter. Введите «y», чтобы указать каждую модель замены отдельно.
После ввода «n» и нажатия клавиши Enter ClockstaR начнет работать. Он распечатает генные деревья на графическом устройстве. Если указанное дерево было корневым, оно также может вывести несколько предупреждений, которые можно смело игнорировать.
Откройте папку clockstar_example_data. Вы найдете файл с именем «example.trees», как указано несколькими шагами выше. Откройте example.trees в текстовом редакторе. Он содержит каждое генное дерево и названия деревьев, соответствующие названиям генных выравниваний. Это должно выглядеть примерно так:
A1 . fasta (( t1 : 0.01504695462 ,( t2 : 0.00987 ...
A2 . fasta (( t1 : 0.01520523401 ,( t2 : 0.01317 ...
A3 . fasta (( t1 : 0.01519309467 ,( t2 : 0.01092 ...
.
.
.Этот файл с деревьями будет использоваться для следующего шага.
Для этого шага необходимо иметь в файле деревья генов, например, полученные на предыдущем шаге.
Откройте R и загрузите ClockstaR, как показано выше. Введите следующий код в командной строке:
clockstar . interactive ()ClockstaR напечатает следующее сообщение:
please drag or type in the path to your gene trees file in NEWICK format :Перетащите файл с генными деревьями в консоль R. Если вы выполнили предыдущий шаг, файл будет называться example.trees. Введите ввод.
В зависимости от установленных вами пакетов ClockstaR может спросить, следует ли ему работать параллельно. Это эффективно для больших наборов данных. Но для данных примера это не будет иметь большого значения, поэтому, если вы видите это сообщение, введите «n», а затем введите Enter:
Packages foreach and doParallel are available for parallel computation
Should we run ClockstaR in parallel (y / n) ? (This is good for large data sets)Clockstar теперь начнет работать. Вывод на экране должен выглядеть примерно так:
[ 1 ] " Calculating sBSDmin distances between all pairs of trees "
[ 1 ] " Estimating tree distances "
[ 1 ] " estimating distances 1 of 11 "
[ 1 ] " estimating distances 2 of 11 "
[ 1 ] " estimating distances 3 of 11 "
[ 1 ] " estimating distances 4 of 11 "
[ 1 ] " estimating distances 5 of 11 "
.
.
.После оценки расстояний между деревьями (описанных в исходной публикации) ClockstaR напечатает следующее сообщение:
" I finished calculating the sBSDmin distances between trees "
The settings for clustering with ClockstaR are :
PAM clustering algorithm
K from 1 to number of data subsets - 1
SEmax criterion to select the optimal k
500 bootstrap replicates
Are these correct ? (y / n)Это настройки алгоритма кластеризации. Они подходят для большинства наборов данных, поэтому в этом примере мы можем ввести «y», а затем ввести. Набрав «n», мы можем изменить эти настройки; более подробную информацию см. в Kaufman and Rousseeuw (2009).
ClockstaR теперь запустит алгоритм кластеризации. В конце он напечатает лучшее количество разделов и спросит, следует ли сохранять результаты в файле pdf:
[ 1 ] " ClockstaR has finished running "
[ 1 ] " The best number of partitions for your data set is: 3 "
Do you wish to save the results in a pdf file ? (y / n)Введите «y», затем введите.
Затем ClockstaR запросит имена выходных файлов:
What should be the name and path of the output file ?Для этого примера введите «example_run» и введите, но можно использовать любое имя.
Теперь откройте папку clockstar_example_data и откройте два файла PDF: example_run_gapstats.pdf и example_run_matrix.pdf.
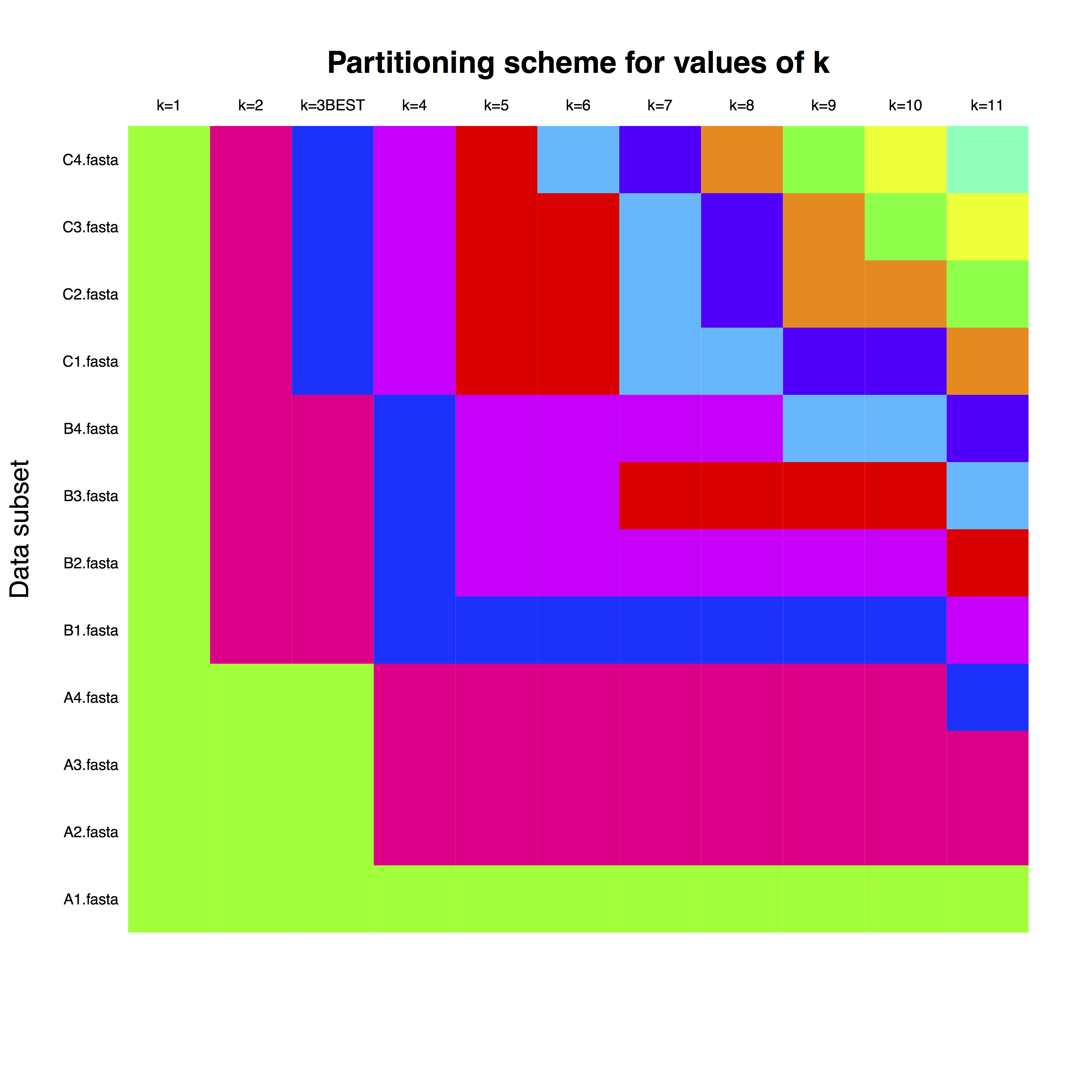
example_run_matrix — это матрица, строки которой соответствуют каждому гену, указанному в файлах FASTA. Столбцы указывают количество разделов, а цвета обозначают назначение каждого гена разделу часов. Например, для k =3, что является наилучшим количеством разделов, можно использовать отдельные разделы часов для генов с буквами A, B и C.
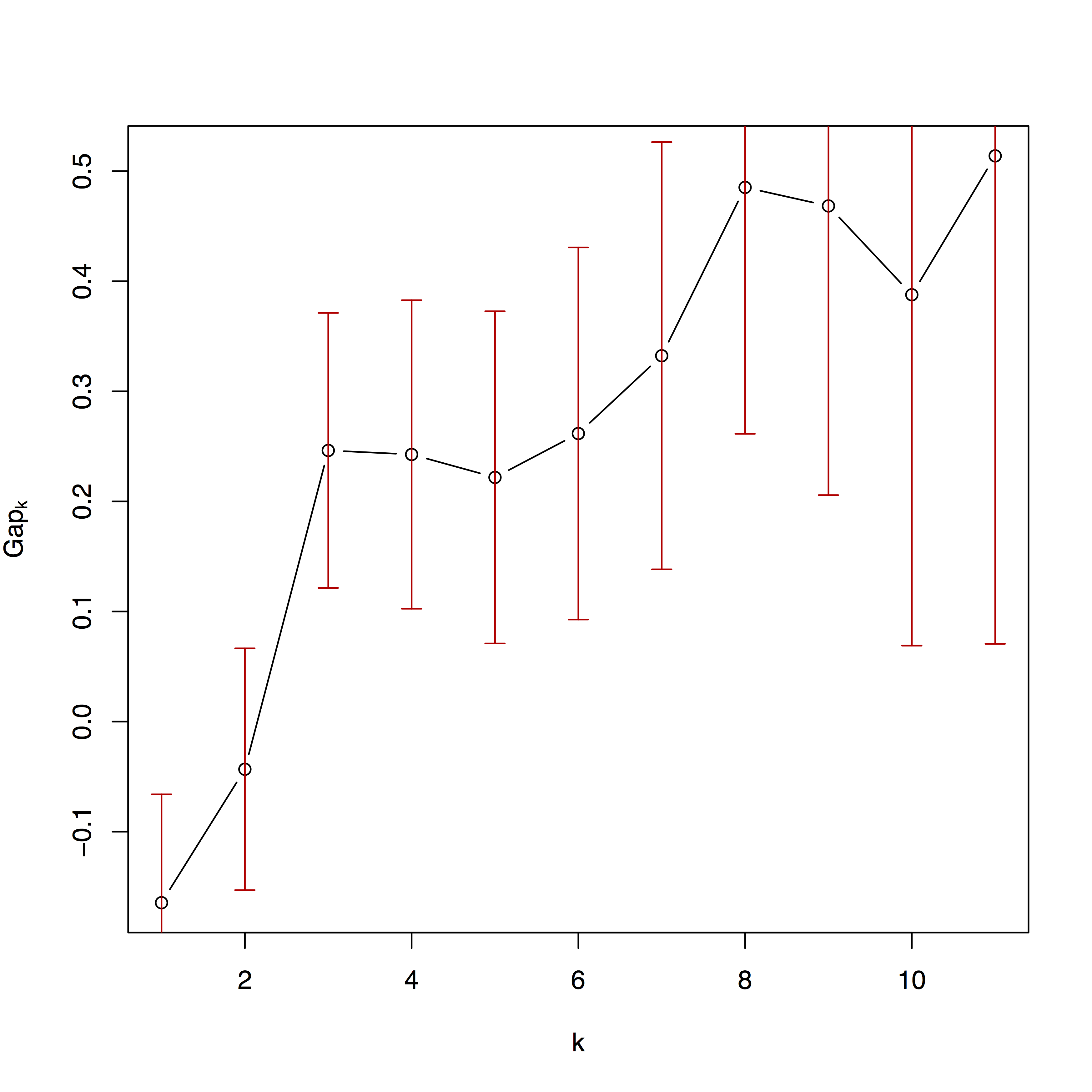
Второй график — это соответствие алгоритмов кластеризации разному количеству разделов. Более подробную информацию можно найти у Кауфмана и Руссеу (2009) и в документации по кластеру пакетов.
ClockstaR можно запускать с другими пользовательскими настройками. Дополнительную информацию см. в документации или по любым вопросам пишите мне на sebastian.duchene[at]sydney.edy.au.
Логотип разработал Джун Тонг.
Дюшен С. и Хо С.Ю. (2014a). Использование нескольких моделей расслабленных часов для оценки временных масштабов эволюции на основе данных о последовательностях ДНК. Молекулярная филогенетика и эволюция (77): 65-70.
Дюшен С., Молак М. и Хо С.Ю. (2014b). ClockstaR: выбор количества моделей с расслабленными часами в молекулярном филогенетическом анализе. Биоинформатика 30 (7): 1017-1019.
Кауфман Л. и Русси П.Дж. (2009). Поиск групп в данных: введение в кластерный анализ (том 344). Джон Уайли и сыновья.


