Это репо содержит:
sepal требует python3 , желательно версии старше 3.5 или равной ей. Чтобы загрузить и установить, откройте терминал и перейдите в каталог, в который вы хотите загрузить sepal , и выполните:
git clone https://github.com/almaan/sepal.git
cd sepal
chmod +x setup.py
./setup.py install
В зависимости от ваших пользовательских привилегий вам, возможно, придется добавить --user в качестве аргумента setup.py . Запуск установки даст вам минимальную установку, необходимую для расчета времени распространения. Однако если вы хотите использовать модули анализа, вам также необходимо установить рекомендуемые пакеты. Для этого просто (в том же каталоге) запустите:
pip install -e " .[full] " опять же, возможно, потребуется включить --user . Кроме того, вам, возможно, придется использовать pip3 , если вы настроили интерфейс python-pip именно так. Если вы используете conda или виртуальные среды, следуйте их рекомендациям по установке пакетов.
При этом должен быть установлен как интерфейс командной строки (CLI), так и стандартный пакет. Чтобы проверить, прошла ли установка успешно, вы можете попробовать выполнить команду:
sepal -h
Который должен напечатать справочное сообщение, связанное с чашелистиком. Если у вас все получилось, вы можете перейти к разделу примеров, чтобы увидеть sepal в действии!
Рекомендуемое использование чашелистика — через интерфейс командной строки. Как моделирование для расчета времени диффузии, так и последующий анализ или проверка результатов можно легко выполнить, набрав sepal с последующим run или analyze . Модуль analyze имеет различные опции: визуализировать результаты ( inspect ), сортировать профили по семействам шаблонов ( family ) или подвергать идентифицированные семейства анализу функционального обогащения ( fea ). Чтобы получить полный список доступных команд, выполните команду sepal module -h , где модуль — это run и analyze . Ниже мы проиллюстрируем, как чашелистник можно использовать для поиска профилей транскрипции с пространственными паттернами.
Мы создадим папку для хранения наших результатов, которая также будет нашим рабочим каталогом. В главном каталоге репо выполните:
cd res
mkdir example
cd exampleОбразец MOB будет использоваться в качестве примера нашего анализа. Начнем с расчета времени диффузии для каждого профиля транскрипции:
sepal run -c ../../data/real/mob.tsv.gz -mo 10 -mc 5 -o . -ar 1Ниже приведен пример (с дополнительным отображением команды справки) того, как это может выглядеть.

Вычислив время диффузии, мы хотим проверить результат, как и в исследовании, мы рассмотрим 20 лучших профилей. Мы можем легко генерировать изображения на основе нашего результата, выполнив команду:
sepal analyze -c ../../data/real/mob.tsv.gz
-r 20200409173043610345-top-diffusion-times.tsv
-ar 1k -o . inspect -ng 20 -nc 5Это будет выглядеть примерно так:

Результатом будет следующее изображение:
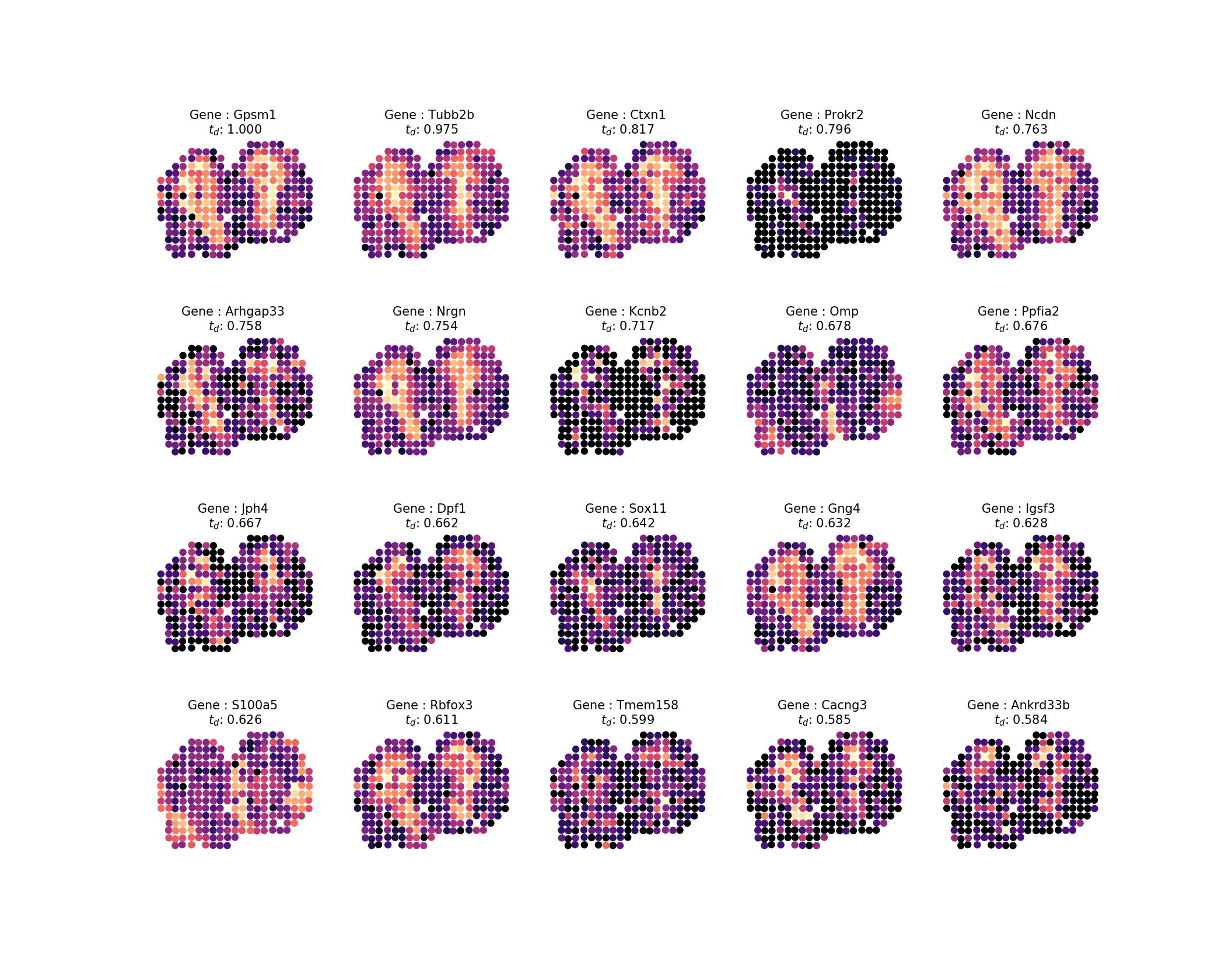
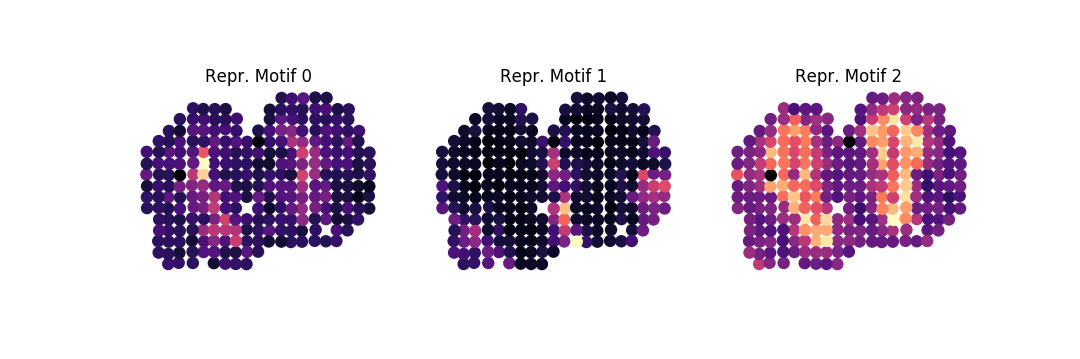
Затем, чтобы отсортировать 100 генов с самым высоким рейтингом в набор семейств паттернов, где 85% дисперсии в наших паттернах должны быть объяснены собственными паттернами, выполните:
sepal analyze -c ../../data/real/mob.tsv.gz
-r 20200409173043610345-top-diffusion-times.tsv
-ar 1k -o . family -ng 100 -nbg 100 -eps 0.85 --plot -nc 3Отсюда мы получаем следующие три репрезентативных мотива для каждой семьи:
Мы можем подвергнуть наши семьи анализу обогащения, запустив:
sepal analyze -c ../../data/real/mob.tsv.gz
-r 20200409173043610345-top-diffusion-times.tsv
-ar 1k -o . fea -fl mob.tsv-family-index.tsv -or " mmusculus "где мы, например, видим, что семейство 2 обогащено несколькими процессами, связанными с функцией, генерацией и регуляцией нейронов:
| семья | родной | имя | p_value | источник | размер_пересечения | |
|---|---|---|---|---|---|---|
| 2 | 2 | ИДТИ: 0007399 | развитие нервной системы | 0,00035977 | ВПЕРЕД:BP | 26 |
| 3 | 2 | ИДТИ: 0050773 | регуляция развития дендритов | 0,000835883 | ВПЕРЕД:BP | 8 |
| 4 | 2 | GO:0048167 | регуляция синаптической пластичности | 0,00196494 | ВПЕРЕД:BP | 8 |
| 5 | 2 | GO:0016358 | развитие дендритов | 0,00217167 | ВПЕРЕД:BP | 9 |
| 6 | 2 | ИДТИ: 0048813 | морфогенез дендритов | 0,00741589 | ВПЕРЕД:BP | 7 |
| 7 | 2 | ИДТИ: 0048814 | регуляция морфогенеза дендритов | 0,00800399 | ВПЕРЕД:BP | 6 |
| 8 | 2 | ИДТИ: 0048666 | развитие нейронов | 0,0114088 | ВПЕРЕД:BP | 16 |
| 9 | 2 | GO:0099004 | сигнальный путь кальмодулинзависимой киназы | 0,0159572 | ВПЕРЕД:BP | 3 |
| 10 | 2 | ИДТИ: 0050804 | модуляция химической синаптической передачи | 0,0341913 | ВПЕРЕД:BP | 10 |
| 11 | 2 | GO:0099177 | регуляция транссинаптической передачи сигналов | 0,0347783 | ВПЕРЕД:BP | 10 |
Конечно, этот анализ ни в коем случае не является исчерпывающим. Это скорее быстрый пример, показывающий, как работает CLI для sepal .
Хотя sepal был разработан как отдельный инструмент, мы также создали его так, чтобы он функционировал как стандартный пакет Python, из которого можно импортировать функции и использовать их в интегрированном рабочем процессе. Чтобы показать, как это можно сделать, мы приведем пример, воспроизводящий анализ меланомы. Позже могут быть добавлены дополнительные примеры.
Входные данные для sepal должны быть в формате n_locations x n_genes , однако, если ваши данные структурированы противоположным образом ( n_genes x n_locations ), просто укажите флаг --transpose при запуске моделирования или анализа, и об этом позаботятся. из.
В настоящее время мы поддерживаем форматы .csv , .tsv и .h5ad . В последнем случае ваш файл должен быть структурирован в соответствии с ЭТИМ форматом. Мы ожидаем, что в ближайшем будущем выйдет релиз от команды scanpy , где будет представлен стандартизированный формат пространственных данных, но до тех пор мы будем использовать вышеупомянутый стандарт.
Все реальные данные, которые мы использовали, являются общедоступными, и их можно найти по следующим ссылкам:
Синтетические данные были получены с помощью:
synthetic/img2cnt.pysynthetic/turing.pysynthetic/ablation.py Все результаты, представленные в исследовании, можно найти в папке res , как для реальных, так и для синтетических данных. Для каждого образца мы соответствующим образом структурировали результаты:
res/sample-name/X-diffusion-times.tsv : время диффузии для всех ранжированных генов.analysis/ : содержит результаты вторичного анализа

