reference_database_creator
bug fix --in-silico-pcr --untrimmed
КРАБЫ ( С чтение Р справочные базы данных для А мпликон- Б Асед С секвенирование) — это универсальная программа, которая создает тщательно подобранные справочные базы данных для метагеномного анализа. Рабочий процесс CRABS состоит из семи модулей: (i) загрузка данных из онлайн-хранилищ; (ii) импортировать загруженные данные в формат CRABS; (iii) извлечение областей ампликона посредством ПЦР-анализа in silico ; (iv) извлекать ампликоны без областей связывания праймеров путем выравнивания со штрих-кодами, извлеченными in silico ; (v) курировать и подбирать локальную базу данных с помощью нескольких параметров фильтрации; (vi) экспортировать локальную базу данных в различные форматы в соответствии с требованиями таксономического классификатора; и (vi) функции постобработки, т.е. визуализации, для изучения и предоставления сводного обзора местной справочной базы данных. Эти семь модулей разделены на восемнадцать функций и описаны ниже. Кроме того, для каждой из восемнадцати функций предоставляется пример кода. Наконец, в конце этого документа README представлено руководство по созданию локальной базы данных по акулам для набора праймеров MiFish-E, в котором представлен пример сценария для справки.
Мы рады сообщить, что в CRABS произошло крупное обновление и редизайн кода на основе отзывов пользователей, которые, как мы надеемся, улучшат удобство работы пользователей при создании вашей собственной локальной справочной базы данных!
Ниже вы найдете список функций и улучшений, добавленных в CRABS v 1.0.0 :
CRABS v 1.0.0 теперь можно загрузить вручную, клонировав этот репозиторий GitHub (подробную информацию см. в разделе 4.1 Ручная установка). Мы обновим контейнер Docker и пакет conda как можно скорее, чтобы облегчить установку новейшей версии.
При использовании CRABS в своих исследовательских проектах цитируйте следующую статью:
[Jeunen, G.-J., Dowle, E., Edgecombe, J., von Ammon, U., Gemmell, N. J., & Cross, H. (2022). crabs—A software program to generate curated reference databases for metabarcoding sequencing data. Molecular Ecology Resources, 00, 1– 14.](https://doi.org/10.1111/1755-0998.13741)
CRABS — это набор инструментов только для командной строки, работающий в типичных средах Unix/Linux и написанный исключительно на Python3. Однако CRABS использует модуль подпроцесса в Python для запуска нескольких команд в синтаксисе bash, чтобы обойти особенности Python и увеличить скорость выполнения. Мы предоставляем три способа установки CRABS. Для самой последней версии CRABS мы рекомендуем установку вручную путем клонирования этого репозитория GitHub и установки 10 зависимостей отдельно (инструкции по установке для всех зависимостей приведены в разделе 4.1 Установка вручную). CRABS также можно установить через Docker и Conda. Оба метода позволяют упростить установку за счет автоматической совместной установки всех зависимостей. Мы стремимся поддерживать контейнер Docker и пакет conda в актуальном состоянии, хотя может возникнуть определенная задержка при обновлении до новейшей версии, особенно для пакета conda. Ниже приведены подробности для всех трех подходов.
Для установки вручную сначала клонируйте репозиторий CRABS. Для этого шага требуется, чтобы GitHub был доступен из командной строки (инструкции по установке GitHub).
git clone https://github.com/gjeunen/reference_database_creator.git
В зависимости от ваших настроек, возможно, CRABS потребуется сделать исполняемым в вашей системе. Этого можно добиться, используя приведенный ниже код.
chmod +x reference_database_creator/crabs
После установки CRABS нам необходимо убедиться, что все зависимости установлены и глобально доступны. Последняя версия CRABS (версия v 1.0.0 ) работает на Python 3.11.7 (или любой версии, совместимой с 3.11.7) и опирается на пять модулей Python, которые могут не входить в стандартную комплектацию Python, а также пять внешних программ. Все зависимости перечислены ниже вместе со ссылкой на инструкции по установке. Номера версий, указанные для каждого модуля и программного обеспечения, соответствуют тем, на которых был разработан CRABS. Хотя можно использовать и совместимые версии каждого из них.
Модули Python:
Внешние программы:
После установки CRABS и всех зависимостей CRABS можно сделать доступным во всей ОС, используя приведенный ниже код.
export PATH="/path/to/crabs/folder:$PATH"
Замените /path/to/crabs/folder фактическим путем к папке репозитория GitHub в ОС, т. е. папкой, созданной во время выполнения приведенной выше команды git clone . Добавление кода export в файл .bash_profile или .bashrc сделает CRABS глобально доступным в любое время.
Docker — это проект с открытым исходным кодом, который позволяет развертывать программные приложения внутри «контейнеров», изолированных от вашего компьютера и запускаемых через операционную систему виртуального хоста под названием Docker Engine. Основное преимущество запуска Docker перед виртуальными машинами заключается в том, что они используют гораздо меньше ресурсов. Эта изоляция означает, что вы можете запускать контейнер Docker в большинстве операционных систем, включая Mac, Windows и Linux. Для использования Docker Desktop вам может потребоваться создать бесплатную учетную запись. Эта ссылка содержит хорошее введение в основы использования Docker. Вот ссылка, которая поможет вам начать работу и ориентироваться в мультивселенной Docker.
Чтобы запустить Crabs на вашем компьютере, нужно выполнить всего два шага. Сначала установите на свой компьютер Docker Desktop, который бесплатен для большинства пользователей. Вот инструкции для Mac ; вот инструкции для компьютеров с Windows и вот инструкции для Linux (поддерживается большинство основных платформ Linux). После установки и запуска Docker Desktop (приложение Desktop должно быть запущено, чтобы вы могли использовать любые команды Docker в командной строке), вам просто нужно «вытащить» наш образ Crabs, и все готово:
docker pull quay.io/swordfish/crabs:0.1.7
Хотя установка приложения Docker проста, поначалу использовать эти приложения может быть немного сложнее. Чтобы помочь вам начать работу, мы предоставили несколько примеров команд, использующих версию crabs для Docker. Эти примеры можно найти в папке docker_intro в этом репозитории . Из этих примеров вы сможете выполнить настройку всей эталонной базы данных и быть готовыми к работе. Мы продолжим расширять эти примеры и тестировать их во многих различных ситуациях. Пожалуйста, задавайте вопросы и оставляйте отзывы на вкладке «Проблемы».
Чтобы установить пакет conda, сначала необходимо установить conda. Подробности смотрите по этой ссылке. Если conda уже установлена, перед установкой CRABS рекомендуется обновить инструмент conda с помощью conda update conda .
После установки conda выполните следующие действия, чтобы установить CRABS и все зависимости. Обязательно вводите команды в том порядке, в котором они указаны ниже.
conda create -n CRABS
conda activate CRABS
conda config --add channels bioconda
conda config --add channels conda-forge
conda install -c bioconda crabs
После того, как вы ввели команду установки, conda обработает запрос (это может занять минуту или около того), а затем отобразит все пакеты и программы, которые будут установлены, и попросит подтвердить это. Введите y чтобы начать установку. После этого CRABS должен быть готов к работе.
Мы протестировали эту установку на системах Mac и Linux. Мы еще не тестировали подсистему Windows для Linux (WSL).
Используйте приведенный ниже код, чтобы проверить, успешно ли установлен CRABS, и откройте справочную информацию.
crabs -hВ справочной информации восемнадцать функций разделены на различные группы, причем в каждой группе вверху указана функция, а внизу — обязательные и необязательные параметры.

CRABS содержит семь модулей, которые включают в себя восемнадцать функций:
Модуль 1: загрузка данных из онлайн-хранилищ
--download-taxonomy : загрузить информацию о таксономии NCBI;--download-bold : загрузить данные последовательности из базы данных Barcode of Life (ЖИРНЫЙ);--download-embl : загрузить данные о последовательностях из Европейского архива нуклеотидов (ENA; EMBL);--download-mitofish : загрузить данные последовательности из базы данных MitoFish;--download-ncbi : загрузить данные последовательности из Национального центра биотехнологической информации (NCBI).Модуль 2: импорт загруженных данных в формат CRABS.
--import : импортировать загруженные последовательности или пользовательские штрих-коды в формат CRABS;--merge : объединить разные файлы в формате CRABS в один файл.Модуль 3. Извлечение областей ампликона с помощью ПЦР-анализа in silico.
--in-silico-pcr : извлекать ампликоны из загруженных данных, находя и удаляя области связывания праймеров.Модуль 4: получение ампликонов без областей связывания праймеров
--pairwise-global-alignment : получить ампликоны без областей связывания праймеров путем выравнивания загруженных последовательностей со штрих-кодами, извлеченными in silico .Модуль 5: курирование и подмножество локальной базы данных с помощью нескольких параметров фильтрации.
--dereplicate : удалить повторяющиеся последовательности;--filter : отбросить последовательности с помощью нескольких параметров фильтрации;--subset : подмножество локальной базы данных, чтобы сохранить или исключить указанные таксономические группы.Модуль 6: экспорт локальной базы данных
--export : экспортировать базу данных в формате CRABS в различные форматы в соответствии с требованиями используемого таксономического классификатора.Модуль 7: Функции постобработки для изучения и предоставления сводного обзора местной справочной базы данных.
--diversity-figure : создает горизонтальную гистограмму, отображающую количество видов и групп последовательностей на указанный уровень, включенных в справочную базу данных;--amplicon-length-figure : создает линейную диаграмму, изображающую распределение длин ампликонов, разделенных по таксономическим группам;--phylogenetic-tree : создает филогенетическое дерево со штрих-кодами из справочной базы данных для целевого списка видов;--amplification-efficiency-figure : создает гистограмму, отображающую несоответствия в областях связывания праймеров;--completeness-table : создает электронную таблицу, содержащую наличие штрих-кодов для таксономических групп.Исходные данные секвенирования могут быть загружены CRABS из четырех онлайн-репозиториев, включая (i) BOLD, (ii) EMBL, (iii) MitoFish и NCBI. Начиная с версии 1.0.0 , загрузка данных из каждого репозитория разделена на отдельные функции. Кроме того, CRABS не форматирует данные автоматически после загрузки, чтобы повысить гибкость и включить отладку в случае сбоя загрузки данных.
Помимо загрузки данных о последовательностях, CRABS также способен загружать информацию о таксономии NCBI, которую CRABS использует для создания таксономической линии для каждой последовательности.
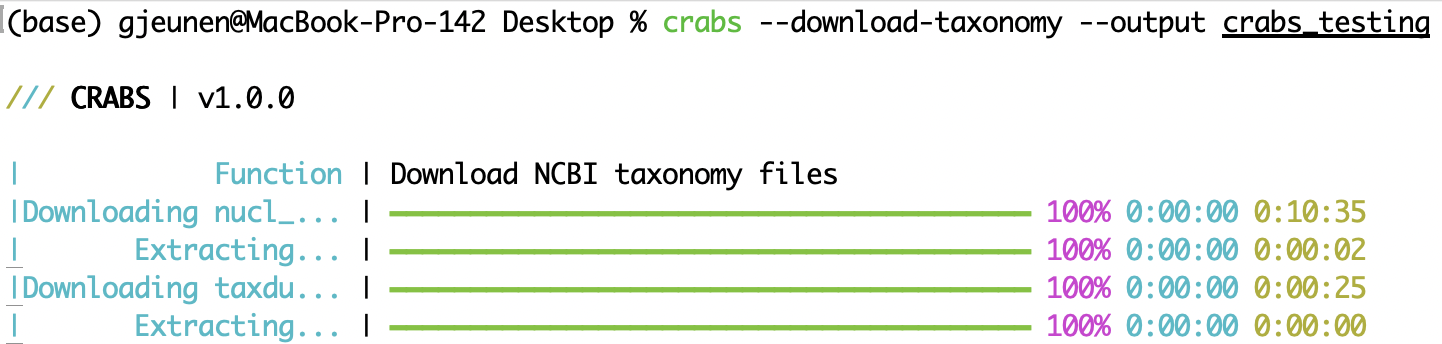
--download-taxonomy Чтобы присвоить таксономическую линию каждой загруженной последовательности в справочной базе данных (см. 5.2 Модуль 2), необходимо загрузить таксономическую информацию. CRABS использует таксономию NCBI и загружает на ваш компьютер три конкретных файла: (i) файл, связывающий инвентарные номера с таксономическими идентификаторами ( nucl_gb.accession2taxid ), (ii) файл, содержащий информацию о филогенетическом имени, связанном с каждым таксономическим идентификатором ( names.dmp ). ) и (iii) файл, содержащий информацию о том, как связаны таксономические идентификаторы ( nodes.dmp ). Выходной каталог для загруженных файлов можно указать с помощью параметра --output . Чтобы исключить файл nucl_gb.accession2taxid или файлы name.dmp и nodes.dmp , можно указать параметр --exclude acc2tax или --exclude taxdump соответственно. Первый код ниже не загружает файлы, поскольку для параметра --exclude указаны и acc2tax , и taxdump . Вторая строка кода загружает все три файла в подкаталог --output crabs_testing . На снимке экрана ниже показано, что выводится на консоль при выполнении этой строки кода.
crabs --download-taxonomy --exclude 'acc2taxid,taxdump'
crabs --download-taxonomy --output crabs_testing
--download-bold Последовательности BOLD загружаются через веб-сайт BOLD. Выходной файл, структурированный как двухстрочный документ fasta, можно указать с помощью параметра --output . Пользователи могут указать, какую таксономическую группу загрузить, используя параметр --taxon . Мы рекомендуем написать простой цикл for (пример приведен ниже), когда пользователи хотят загрузить несколько таксономических групп, тем самым ограничивая объем данных, загружаемых из BOLD для каждого экземпляра. Однако, если интерес представляет только ограниченное число таксономических групп, названия таксономических групп также можно разделить | (пример приведен ниже). Мы также рекомендуем пользователям проверить, указано ли название загружаемой таксономической группы в архиве BOLD или необходимо использовать альтернативные названия. Например, если указать --taxon Chondrichthyes не будут загружены все последовательности хрящевых рыб, выделенные ЖИРНЫМ шрифтом, поскольку это имя класса не указано ЖИРНЫМ шрифтом. В этом случае пользователям лучше использовать --taxon Elasmobranchii . Пользователи также могут указать ограничение загрузки определенным генетическим маркером, указав параметр --marker . Если интерес представляют несколько генетических маркеров, имена маркеров должны быть разделены знаком | . Четыре основных маркера штрих-кодирования ДНК, выделенные жирным шрифтом, — это COI-5P , ITS , matK и rbcL . Ввод параметра --marker чувствителен к регистру.
Рекомендуемый подход: простой цикл for для загрузки данных из BOLD для нескольких таксономических групп (рекомендуемый подход). Код ниже сначала загружает данные для Elasmobranchii, а затем последовательности, присвоенные Mammalia. Загруженные данные будут записаны в подкаталог --output crabs_testing и помещены в два отдельных файла с указанием, какие данные принадлежат к какой таксономической группе, т. е. crabs_testing/bold_Elasmobranchii.fasta и crabs_testing/bold_Mammalia.fasta .
for taxon in Elasmobranchii Mammalia; do crabs --download-bold --taxon ${taxon} --output crabs_testing/bold_${taxon}.fasta; done

Альтернативный вариант: помимо рекомендуемого цикла for, можно указать несколько названий таксонов одновременно, разделив их с помощью | .
crabs --download-bold --taxon 'Elasmobranchii|Mammalia' --output crabs_testing/bold_elasmobranchii_mammalia.fasta
--download-embl Последовательности из EMBL загружаются через FTP-сайт ENA. Файлы EMBL сначала будут загружены в формате «.fasta.gz» и автоматически разархивированы после завершения загрузки. Эта база данных не обеспечивает такой гибкости в отношении выборочной загрузки по сравнению с BOLD или NCBI. Скорее, данные EMBL структурированы по 15 налоговым подразделениям, которые можно загрузить отдельно. Налоговое подразделение для загрузки можно указать с помощью параметра --taxon . Поскольку каждое налоговое подразделение разделено на несколько файлов, после имени указывается * для загрузки всех файлов. Пользователи также могут загрузить конкретный файл, написав его имя полностью. Список всех 15 вариантов налогового разделения представлен ниже. Выходной каталог и имя файла можно указать с помощью параметра --output .
Перечень налоговых подразделений:
crabs --download-embl --taxon 'mam*' --output crabs_testing/embl_mam.fasta
--download-mitofish CRABS также может загрузить базу данных MitoFish. Эта база данных представляет собой один двухстрочный фаст-файл. Выходной каталог и имя файла можно указать с помощью параметра --output .
crabs --download-mitofish --output crabs_testing/mitofish.fasta

--download-ncbi Последовательности из базы данных NCBI загружаются с помощью утилит программирования Entrez. NCBI позволяет загружать данные из различных баз данных, которые пользователи могут указать с помощью параметра --database . Для большинства пользователей база данных --database nucleotide будет наиболее подходящей для создания локальной справочной базы данных.
Чтобы указать данные для загрузки из NCBI, пользователи обеспечивают поиск по параметру --query . Создание хорошего поиска NCBI может быть трудным. Хороший способ создать поисковый запрос — использовать окно поиска на веб-странице NCBI. По этой ссылке сначала выполните первоначальный поиск и нажмите Enter. Это приведет вас на страницу результатов, где вы сможете уточнить свой поиск. На снимке экрана ниже мы еще больше усовершенствовали поиск, ограничив длину последовательности 100–25 000 п.н. и включив только митохондриальные последовательности. Пользователи могут скопировать и вставить текст в поле «Сведения о поиске» на веб-сайте и указать его в кавычках для параметра --query . Еще одним преимуществом использования окна поиска на веб-странице NCBI является то, что на веб-странице будет отображаться количество последовательностей, соответствующих вашему поисковому запросу, что должно соответствовать количеству последовательностей, сообщенных CRABS. На этой веб-странице представлено еще одно краткое руководство по использованию функции поиска на веб-странице NCBI, написанное нашей командой для получения дополнительной информации.
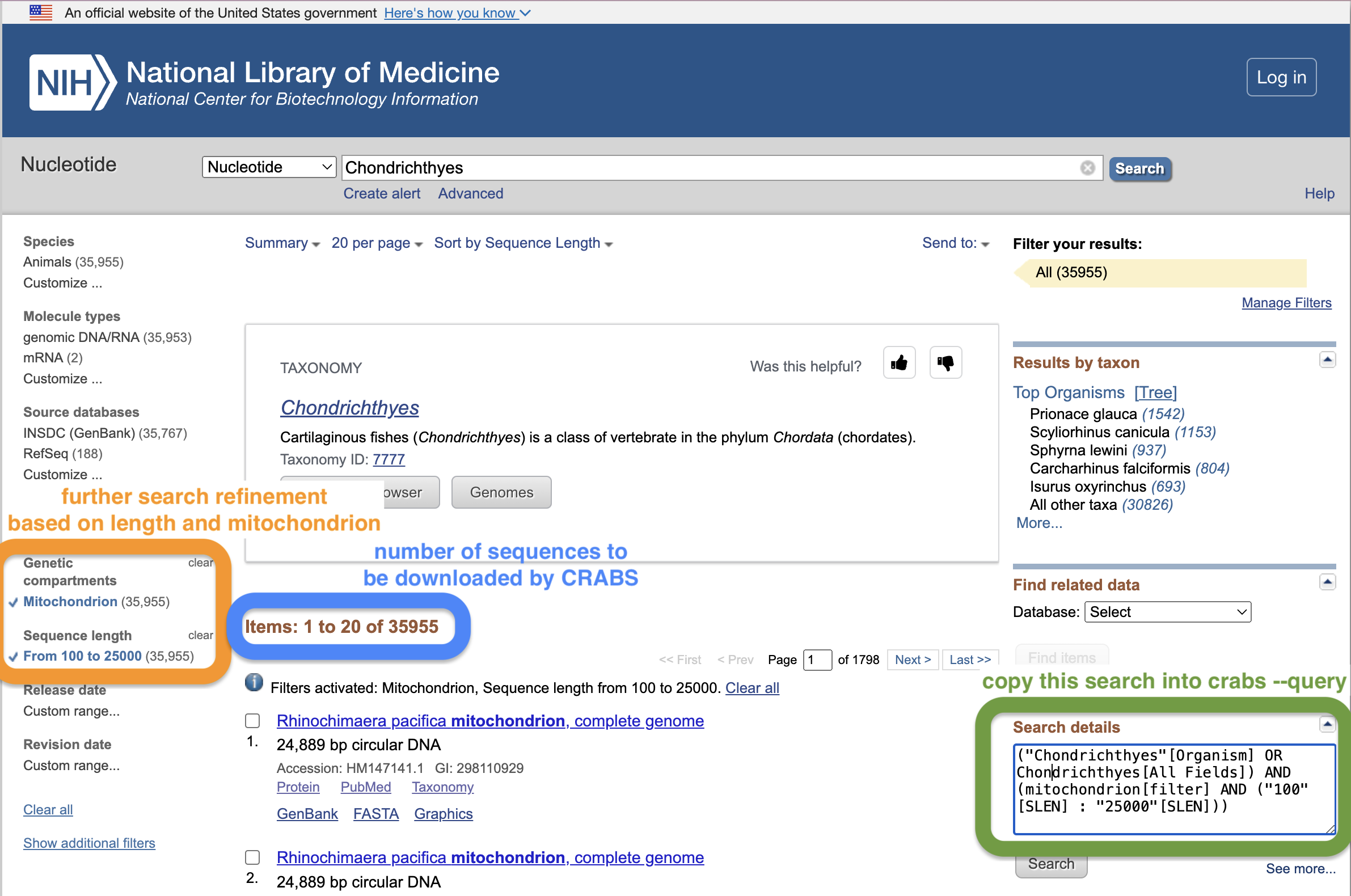
Помимо поискового запроса ( --query ), пользователи могут дополнительно ограничить поисковый запрос, загрузив данные последовательности для списка видов с помощью параметра --species . Параметр --species принимает либо входную строку названий видов, разделенных знаком + , либо входной файл .txt с одним названием вида в каждой строке документа. Параметр --batchsize предоставляет пользователям возможность загружать последовательности пакетами по N с веб-сайта NCBI. По умолчанию этот параметр равен 5000. Не рекомендуется увеличивать это значение выше 5000, поскольку серверы NCBI, скорее всего, отключат загрузку, если одновременно будет загружено слишком много последовательностей. Параметр --email позволяет пользователям указать свой адрес электронной почты, который необходим для доступа к серверам NCBI. Наконец, выходной каталог и имя файла можно указать с помощью параметра --output .
crabs --download-ncbi --query '("Chondrichthyes"[Organism] OR Chondrichthyes[All Fields]) AND (mitochondrion[filter] AND ("100"[SLEN] : "25000"[SLEN]))' --output crabs_testing/ncbi_chondrichthyes.fasta --email [email protected] --database nucleotide

--import После загрузки данных из онлайн-хранилищ файлы необходимо будет импортировать в CRABS с помощью функции --import . Формат CRABS представляет собой одну строку, разделенную табуляцией для каждой последовательности, содержащую всю информацию, включая (i) идентификатор последовательности, (ii) таксономическое название, проанализированное из первоначальной загрузки, (iii) идентификационный номер таксона NCBI, (iv) таксономическое происхождение в соответствии с таксономией NCBI. и (v) последовательность. CRABS попытается получить инвентарный номер NCBI для каждой последовательности в качестве идентификатора последовательности. Если последовательность не содержит инвентарного номера, т. е. она не депонирована в NCBI, CRABS сгенерирует уникальные идентификаторы последовательностей, используя следующий формат: crabs_*[num]*_taxonomic_name . Формат входного документа указывается с помощью параметра --import-format и указывает имя репозитория, из которого были загружены данные, т. е. BOLD , EMBL , MITOFISH или NCBI . Таксономическая линия, создаваемая CRABS, основана на таксономии NCBI, и CRABS требует трех файлов, загруженных с помощью функции --download-taxonomy , то есть --names , --nodes и --acc2tax . Начиная с версии 1.0.0 , CRABS способен разрешать синонимы и непринятые имена для включения большего количества последовательностей и разнообразия в локальную справочную базу данных. Таксономические ранги, которые будут включены в таксономическую линию, можно указать с помощью параметров --ranks . Хотя можно включить любой таксономический ранг, мы рекомендуем использовать следующие входные данные для включения всей необходимой информации для большинства таксономических классификаторов --ranks 'superkingdom;phylum;class;order;family;genus;species' . Выходной файл можно указать с помощью параметра --output и представляет собой простой файл .txt. В окне терминала CRABS печатает результаты количества импортированных последовательностей, а также любые последовательности, для которых не удалось создать таксономическую линию.
crabs --import --import-format bold --input crabs_testing/bold_Elasmobranchii.fasta --names crabs_testing/names.dmp --nodes crabs_testing/nodes.dmp --acc2tax crabs_testing/nucl_gb.accession2taxid --output crabs_testing/crabs_bold.txt --ranks 'superkingdom;phylum;class;order;family;genus;species'

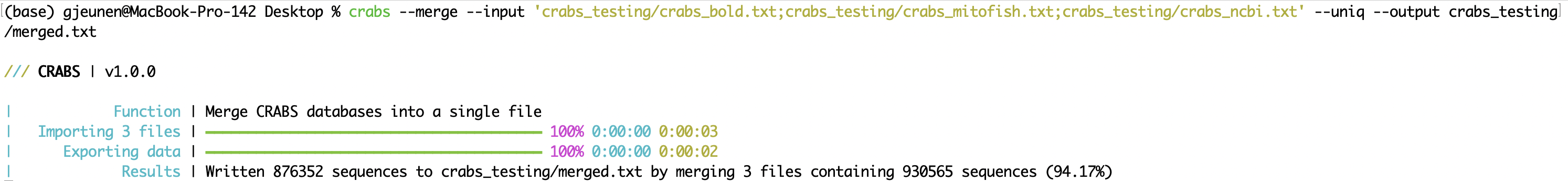
--merge Когда данные последовательности загружаются из нескольких онлайн-репозиториев, файлы можно объединить в один файл после импорта (см. 5.2.1 --import ) с помощью функции --merge . Входные файлы для объединения можно ввести с помощью параметра --input , разделив файлы ; . Вполне возможно, что эпизод был загружен несколько раз при размещении в различных онлайн-репозиториях. Использование параметра --uniq сохраняет только одну версию каждого номера доступа. Выходной файл можно указать с помощью параметра --output . В окне терминала CRABS печатает результаты количества объединенных последовательностей, а также количества сохраненных последовательностей при использовании параметра --uniq .
crabs --merge --input 'crabs_testing/crabs_bold.txt;crabs_testing/crabs_mitofish.txt;crabs_testing/crabs_ncbi.txt' --uniq --output crabs_testing/merged.txt
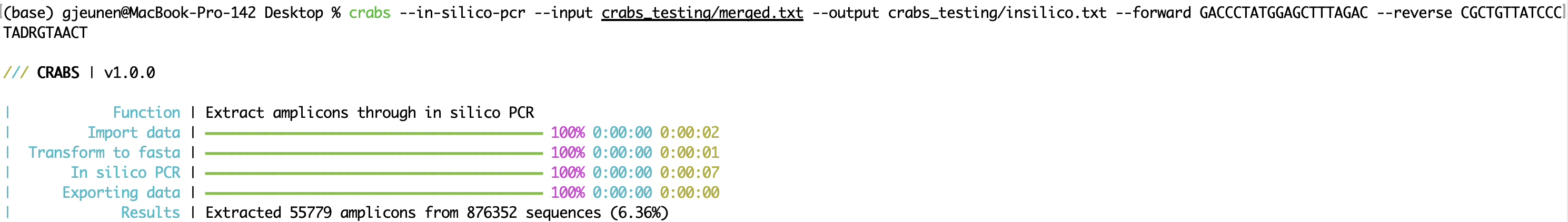
CRABS извлекает область ампликона из набора праймеров путем проведения ПЦР in silico (функция: --in-silico-pcr ). CRABS использует Cutadapt v 4.4 для PCR in silico , чтобы увеличить скорость выполнения традиционного кода Python. Имена входных и выходных файлов можно указать с помощью параметров --input и --output соответственно. И прямой, и обратный праймеры должны быть предоставлены в направлении 5'-3' с использованием параметров ' --forward ' и ' --reverse ' соответственно. КРАБЫ будут обратно дополнять обратный праймер. Начиная с версии 1.0.0 , CRABS способен сохранять штрих-коды в обоих направлениях с использованием одного ПЦР-анализа in silico . Следовательно, не проводится этап обратной комплементации и повторный запуск ПЦР in silico , что значительно увеличивает скорость выполнения. Чтобы сохранить последовательности, для которых не удалось найти области связывания праймеров, для параметра --untrimmed можно указать выходной файл. Максимально допустимое количество несоответствий, обнаруженных в областях связывания праймеров, можно указать с помощью параметра --mismatch со значением по умолчанию, равным 4. Наконец, анализ ПЦР in silico может быть многопоточным в CRABS. По умолчанию используется максимальное количество потоков, но пользователи могут указать количество используемых потоков с помощью параметра --threads .
crabs --in-silico-pcr --input crabs_testing/merged.txt --output crabs_testing/insilico.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT
Обычной практикой является удаление областей связывания праймеров из эталонных последовательностей при их размещении в онлайн-базе данных. Следовательно, если эталонная последовательность была создана с использованием того же прямого и/или обратного праймера, который был найден в функции --in-silico-pcr , функция --in-silico-pcr не сможет восстановить область ампликона эталонная последовательность. Чтобы учесть эту возможность, CRABS имеет возможность запустить парное глобальное выравнивание, реализованное с использованием VSEARCH v 2.16.0 , для извлечения областей ампликона, для которых эталонная последовательность не содержит полных областей прямого и обратного связывания праймеров. Для этого функция --pairwise-global-alignment принимает исходно загруженный файл базы данных, используя параметр --input . База данных, в которой производится поиск, представляет собой выходной файл --in-silico-pcr и может быть указана с помощью параметра --amplicons . Выходной файл можно указать с помощью параметра --output . Последовательности праймеров, используемые только для расчета длины пары оснований, могут быть установлены с помощью параметров --forward и --reverse . Поскольку выполнение функции --pairwise-global-alignment в больших базах данных может занять много времени, длину последовательности можно ограничить, чтобы ускорить процесс, с помощью параметра --size-select . Минимальный процент идентичности и покрытия запросов можно указать с помощью параметров --percent-identity и --coverage соответственно. --percent-identity должен быть указан в виде процентного значения от 0 до 1 (например, 95% = 0,95), а --coverage должен быть указан в виде процентного значения от 0 до 100 (например, 95% = 95). По умолчанию функция --pairwise-global-alignment ограничена сохранением последовательностей, в которых последовательности праймеров не полностью присутствуют в эталонной последовательности (выравнивание начинается или заканчивается в пределах длины прямого или обратного праймера). Если указан параметр --all-start-positions , положительные совпадения будут включены, если выравнивание обнаружено за пределами диапазона областей связывания праймера (пропускается функцией --in-silico-pcr из-за слишком большого количества несоответствий в область связывания праймера). Мы не рекомендуем использовать --all-start-positions , поскольку очень маловероятно, что штрих-код будет амплифицирован с использованием указанного набора праймеров функции --in-silico-pcr , если в праймере присутствует более 4 несоответствий. связывающие области.
crabs --pairwise-global-alignment --input crabs_testing/merged.txt --amplicons crabs_testing/insilico.txt --output crabs_testing/aligned.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT --size-select 10000 --percent-identity 0.95 --coverage 0.95
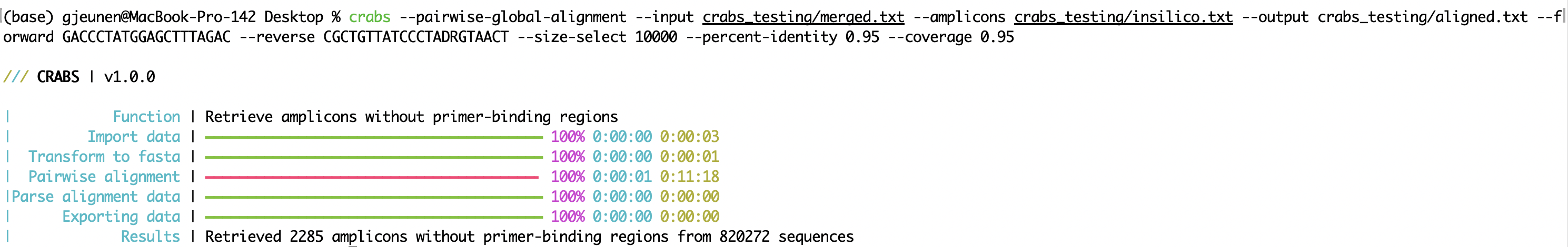
--pairwise-global-alignment Выполнение функции --pairwise-global-alignment может занять значительное время, когда CRABS обрабатывает большие файлы последовательностей, даже если многопоточность поддерживается. Начиная с обновления CRABS v 1.0.0 , от --import до --export используется идентичная файловая структура, что позволяет выполнять функции в любом порядке. Хотя мы по-прежнему рекомендуем следовать порядку рабочего процесса CRABS, функцию --pairwise-global-alignment можно значительно ускорить, выполняя функции --dereplicate и --filter до функции --in-silico-pcr . Выполняя эти шаги курирования до --in-silico-pcr , количество последовательностей, которые необходимо обработать CRABS для функции --pairwise-global-alignment будет значительно уменьшено.
ПРИМЕЧАНИЕ 1 : при выполнении функции --filter перед --in-silico-pcr обязательно опустите любые параметры, которые непосредственно влияют на последовательность, поскольку --filter будет основывать это на всей последовательности, а не на извлеченном ампликоне. . Следовательно, опустите следующие параметры: --minimum-length , --maximum-length , --maximum-n .
ПРИМЕЧАНИЕ 2 : при выполнении функций --dereplicate и --filter до --in-silico-pcr было бы целесообразно снова запустить обе функции после --pairwise-global-alignment , так как теперь базу данных можно будет дополнительно курировать. что ампликоны извлечены.
После того, как все потенциальные штрих-коды для набора праймеров будут извлечены с помощью функций --in-silico-pcr и --pairwise-global-alignment , локальная справочная база данных может подвергаться дальнейшему курированию и подмножеству в CRABS с использованием различных функций, включая --dereplicate , --filter и --subset .
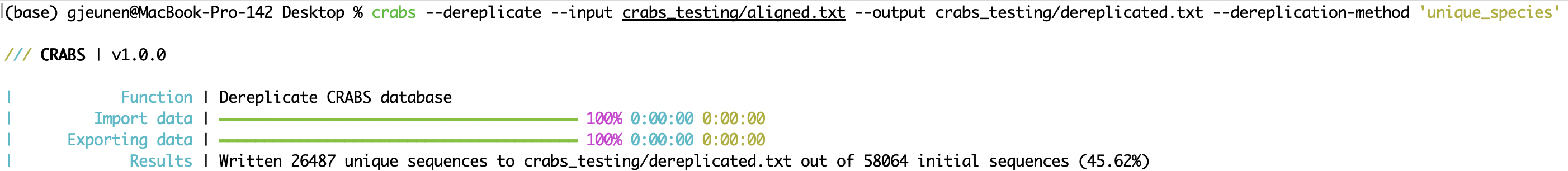
--dereplicate Первый метод курирования — отменить репликацию локальной справочной базы данных с помощью функции --dereplicate . Вполне возможно, что для некоторых таксонов на данный момент в местной справочной базе данных содержится несколько идентичных штрих-кодов. Это может произойти, когда разные исследовательские группы депонировали идентичные последовательности или если внутривидовые различия между последовательностями таксона не содержатся в извлеченном штрих-коде. Лучше всего удалить эти идентичные эталонные штрих-коды, чтобы ускорить присвоение таксономии, а также улучшить результаты присвоения таксономии (особенно для таксономических классификаторов, предоставляющих ограниченное количество результатов, например, BLAST).
Входные и выходные файлы можно указать с помощью параметров --input и --output соответственно. CRABS предлагает три метода дерепликации, которые можно указать с помощью параметра --dereplication-method , в том числе:
crabs --dereplicate --input crabs_testing/aligned.txt --output crabs_testing/dereplicated.txt --dereplication-method 'unique_species'
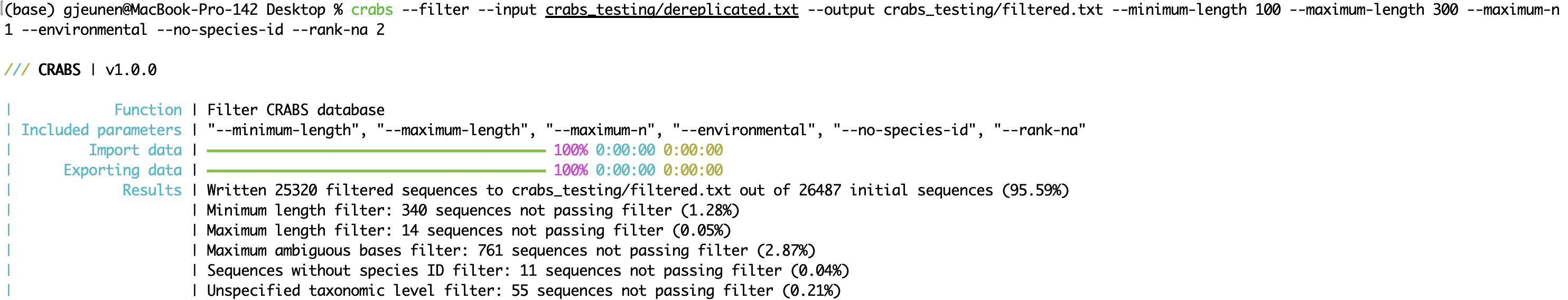
--filter Второй метод курирования — фильтрация локальной справочной базы данных по различным параметрам с помощью функции --filter . Входные и выходные файлы можно указать с помощью параметров --input и --output соответственно. С версии v 1.0.0 . CRABS включает в себя фильтрацию по шести параметрам, в том числе:
--minimum-length : минимальная длина последовательности, при которой ампликон будет храниться в базе данных;--maximum-length : максимальная длина последовательности ампликона, сохраняемая в базе данных;--maximum-n : отбросить ампликоны с N или более неоднозначными основаниями ( N );--environmental : удалить последовательности окружающей среды из базы данных;--no-species-id : отбросить последовательности, для которых нет названия вида;--rank-na : отбросить последовательности с N или более неуказанными таксономическими уровнями. crabs --filter --input crabs_testing/dereplicated.txt --output crabs_testing/filtered.txt --minimum-length 100 --maximum-length 300 --maximum-n 1 --environmental --no-species-id --rank-na 2
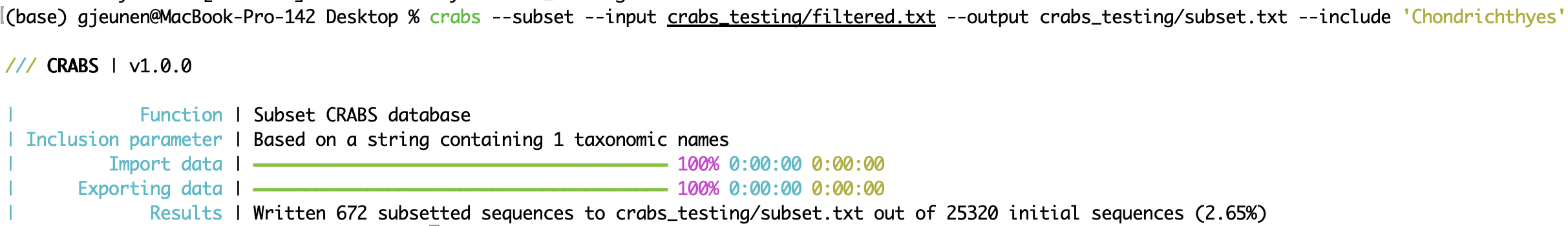
--subset Третий и последний метод курирования, включенный в CRABS, заключается в подмножестве локальной справочной базы данных для включения (параметр: --include ) или исключения (параметр: --exclude ) конкретных таксонов с помощью функции --subset . Эта функция позволяет удалять эталонные штрих-коды из таксономических групп, не представляющих интерес для вопроса исследования. Эти таксономические группы могли быть включены в местную справочную базу данных из-за потенциальной неспецифической амплификации набора праймеров. Другой вариант использования --subset — удаление известных ошибочных последовательностей.
Для таксономических классификаторов, основанных на машинном обучении (IDTAXA) или расстоянии k-mer (SINTAX), может быть полезно разбить справочную базу данных на части, включив только таксоны, которые, как известно, встречаются в регионе, где были взяты образцы, и исключить близкородственные виды, о которых известно, что они не известны. произойти в регионе для увеличения полученного таксономического разрешения этих классификаторов и получения улучшенных результатов присвоения таксономии.
Входные и выходные файлы можно указать с помощью параметров --input и --output соответственно. Параметры --include и --exclude могут принимать либо список таксонов, разделенных ; или файл .txt, содержащий по одному названию таксона в каждой строке.
crabs --subset --input crabs_testing/filtered.txt --output crabs_testing/subset.txt --include 'Chondrichthyes'
После завершения создания справочной базы данных ее можно экспортировать в различные форматы для соответствия спецификациям, требуемым большинством программных инструментов, определяющих таксономию метагеномных данных. Входные и выходные файлы можно указать с помощью параметров --input и --output соответственно. Начиная с версии 1.0.0 , CRABS включает форматирование справочной базы данных для шести различных классификаторов (параметр: --export-format ), включая:
--export-format 'sintax' : классификатор SINTAX включен в USEARCH и VSEARCH;--export-format 'rdp' : Классификатор RDP — это отдельная программа, широко используемая в исследованиях микробиома;--export-format 'qiime-fasta' и --export-format 'qiime-text' : могут использоваться для присвоения таксономического идентификатора в QIIME и QIIME2;--export-format 'dada2-species' и --export-format 'dada2-taxonomy' : могут использоваться для присвоения таксономического идентификатора в DADA2;--export-format 'idt-fasta' и --export-format 'idt-text' : классификатор IDTAXA — это алгоритм машинного обучения, включенный в пакет DECIPHER R;--export-format 'blast-notax' : создает локальную справочную базу данных BLAST для Blastn и Megablast, где выход не предоставляет таксономический идентификатор, но перечисляет номер доступа;--export-format 'blast-tax' : создает локальную справочную базу данных Blast для Blastn и Megablast, где вывод обеспечивает как таксономический идентификатор, так и номер доступа. crabs --export --input crabs_testing/subset.txt --output crabs_testing/BLAST_TAX_CHONDRICHTHYES --export-format 'blast-tax'

При экспорте локальной справочной базы данных в один формат (за исключением классификаторов, где эталонная база данных разделена по нескольким файлам, то есть QIIME, DADA2, IDTAXA) будет достаточно для большинства пользователей, может быть записано для экспорта локального Справочная база данных с несколькими форматами, если пользователи хотели бы сравнить результаты между различными таксономическими классификаторами. Ниже приведен пример для экспорта локальной справочной базы данных в форматах Sintax, RDP и Idtaxa.
for format in sintax.fasta rdp.fasta idt-fasta.fasta idt-text.txt; do crabs --export --input crabs_testing/subset.txt --output crabs_testing/chondrichthyes_${format} --export-format ${format%%.*}; done

Как только эталонная база данных завершена, крабы могут выполнять пять функций после обработки для изучения и предоставления сводного обзора локальной справочной базы данных, включая (i) --diversity-figure , (ii) --amplicon-length-figure . iii) --phylogenetic-tree , (IV) --amplification-efficiency-figure и (v) --completeness-table .
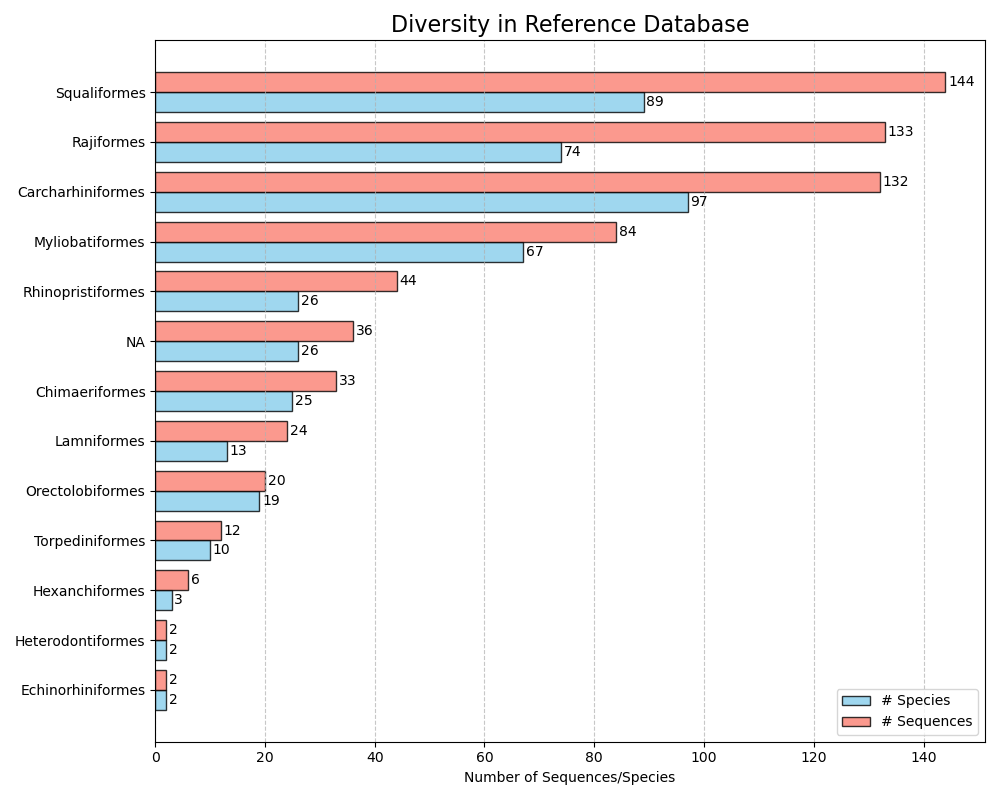
--diversity-figure Функция --diversity-figure создает горизонтальный планшет с количеством видов (синим цветом) и количеством последовательностей (в оранжевом) на каждую таксономическую группу в эталонной базе данных. Пользователь может указать таксономический ранг, чтобы разделить эталонную базу данных с параметром --tax-level . Уровень налога -это количество ранга, в котором он появился во время функции --import . Например, если --ranks 'superkingdom;phylum;class;order;family;genus;species' был использован во время расщепления --import на основе суперкинга, потребовалось бы --tax-level 1 , phylum = --tax-level 2 , Class = --tax-level 3 и т. Д. Входной файл в формате крабов может быть указан с использованием параметра --input . Рисунок в формате .png будет записан в выходной файл, который может быть указан с использованием параметра --output .
crabs --diversity-figure --input crabs_testing/subset.txt --output crabs_testing/diversity-figure.png --tax-level 4
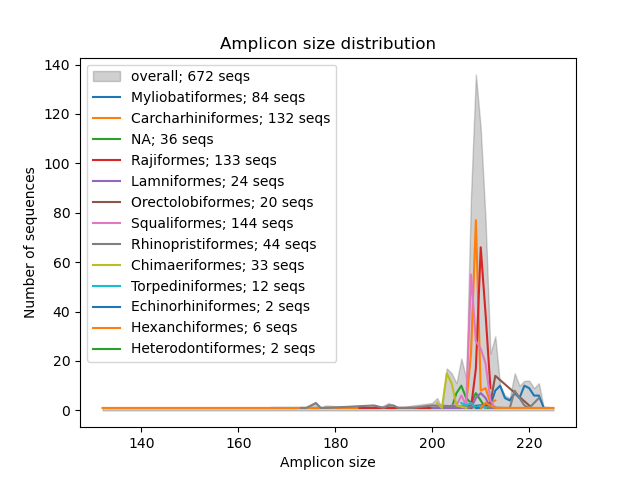
--amplicon-length-figure Функция --amplicon-length-figure создает линейный график, отображающий диапазон длины Amplicon. Общий диапазон длины Amplicon во всех последовательностях в эталонной базе данных отображается в затененном сером цвете, в то время как результаты разделены на таксономическую группу (параметр: --tax-level ) накладываются цветными линиями. Кроме того, легенда отображает количество последовательностей, назначенных каждой из таксономических групп, и общее количество последовательностей в эталонной базе данных. Входной файл в формате крабов может быть указан с использованием параметра --input . Рисунок в формате .png будет записан в выходной файл, который может быть указан с использованием параметра --output .
crabs --amplicon-length-figure --input crabs_testing/subset.txt --output crabs_testing/amplicon-length-figure.png --tax-level 4
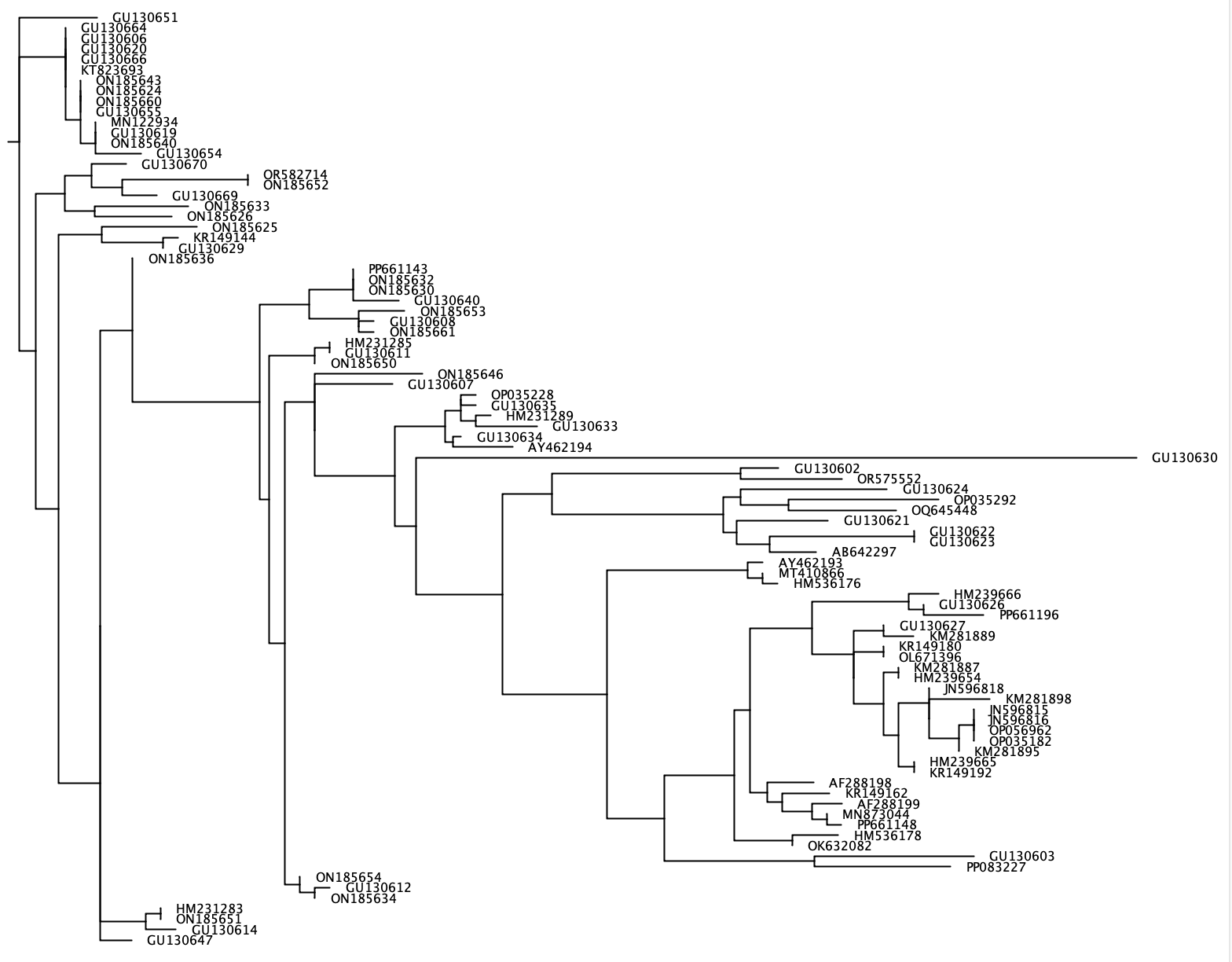
--phylogenetic-tree Функция --phylogenetic-tree будет генерировать филогенетическое дерево для списка интересующих видов. Этот список интересующих видов может быть импортирован с использованием параметра --species и состоит либо из входной строки, разделенной + или файла .txt с одним названием одного вида на каждой строке. Для каждого вида, представляющего интерес, последовательности будут извлечены из эталонной базы данных, в которой есть пользовательский таксономический ранг (параметр: --tax-level ) с интересующим видом. Крабы генерируют выравнивание всех извлеченных последовательностей с использованием ClustalW2 V 2.1 и генерируют филогенетическое дерево с соседством с использованием Fasttree. Это филогенетическое дерево в формате Newick будет записано в выходной файл с использованием параметра --output и может быть визуализировано в программах, таких как Figtree или Geneious. Поскольку для каждого интересующего вида будет генерироваться отдельное филогенетическое дерево, --output общий имя файла, в то время как точный выходной файл будет содержать это общее имя, за которым следует '_species_name.tree'.
crabs --phylogenetic-tree --input crabs_testing/subset.txt --output crabs_testing/phylo --tax-level 4 --species 'Carcharodon carcharias+Squalus acanthias'
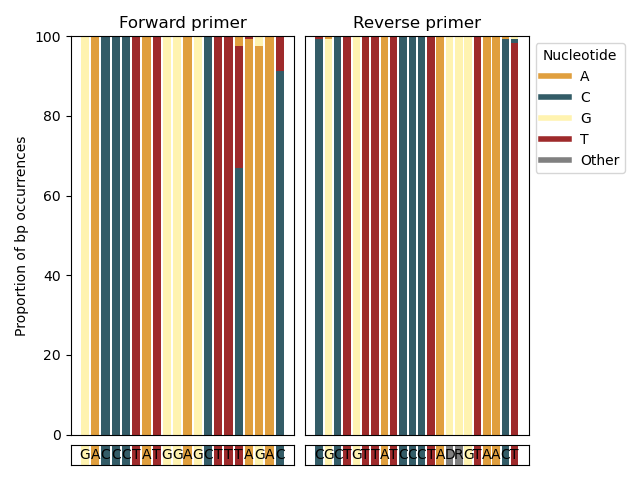
--amplification-efficiency-figure Функция --amplification-efficiency-figure будет создавать гистограмму, отображая долю возникновения пары оснований в области связывания праймеров для таксономической группы, определенной пользователем, и тем самым визуализируя места в направленных и обратных областях связывания праймеров, где несоответствие несоответствия может происходить в таксономической интересной группе, что может влиять на эффективность усиления. Функция --amplification-efficiency-figure принимает окончательную ориентированную базу данных, форматированную крабами в качестве входного параметра с использованием параметра --amplicons . Чтобы найти информацию в области связывания праймеров для каждой последовательности во входном файле, первоначально загруженные последовательности после импорта должны быть предоставлены с использованием параметра --input . Передовые и обратные последовательности праймеров (в направлении 5 ' -3') предоставляются с использованием параметров --forward и --reverse . Название таксономической группы интересов может быть предоставлено с использованием параметра --tax-group и может быть установлено на любом таксономическом уровне, который включен в входной файл. Наконец, рисунок в формате .png будет записан в выходной файл, указанный параметром --output .
crabs --amplification-efficiency-figure --input crabs_testing/merged.txt --amplicons crabs_testing/subset.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT --output crabs_testing/amplification-efficiency.png --tax-group Carcharhiniformes
--completeness-table Функция --completeness-table выведет таблицу, определенную вкладками (параметр: --output ) с информацией о списке интересующих видов. Этот список интересующих видов может быть импортирован с использованием параметра --species и состоит либо из входной строки, разделенной + или файла .txt с одним названием одного вида на каждой строке. Таксономическая линия будет генерироваться для каждого интересующего вида с использованием файлов « names.dmp » и « Nodes.dmp », загруженные с использованием функции --download-taxonomy с использованием параметров --names и --nodes соответственно. В таблице вывода будет 10 столбцов, предоставляющих следующую информацию:
crabs --completeness-table --input crabs_testing/subset.txt --output crabs_testing/completeness.txt --names crabs_testing/names.dmp --nodes crabs_testing/nodes.dmp --species 'Carcharodon carcharias+Squalus acanthias'
crabs --version v 1.0.6 : Исправление ошибок -> Улучшенное анализ жирных жирных заголовков во время --import .crabs --version v 1.0.5 : Исправление ошибок -> Добавлен ограничение длины в идентификатор SEQ при создании баз данных взрыва, как это необходимо для программного обеспечения Blast+.crabs --version v 1.0.4 : добавленная информация-> предоставлена правильная информация о входе значения для --pairwise-global-alignment --coverage --percent-identity .crabs --version v 1.0.3 : Исправление ошибок -> Проверка ответа сервера NCBI за 3 раза до прерывания анализа.crabs --version v 1.0.2 : Исправление ошибки -> Способен отчитываться, когда 0 последовательностей возвращаются после анализа.crabs --version v 1.0.1 : Исправление ошибок -> Успешное строительство запроса NCBI с использованием параметра --species .

