nucleotide transformer
1.0.0
Добро пожаловать в репозиторий InstaDeep на Github, где представлены:
Мы очень рады открыть исходный код этих работ и предоставить сообществу доступ к коду и предварительно обученным весам для этих девяти языковых моделей геномики и двух моделей сегментации. Модели из проекта nucleotide transformer были разработаны в сотрудничестве с Nvidia и TUM, а также прошли обучение на узлах DGX A100 на Кембридже-1. Модель из проекта Агронуклеотидного nucleotide transformer была разработана совместно с Google и обучена на ускорителях TPU-v4.
В целом, наши работы дают новые идеи, связанные с предварительной тренировкой и применением базовых моделей языка, а также обучением моделей, использующих их в качестве основного кодировщика, для геномики с широкими возможностями их применения в этой области.
В этом репозитории вы найдете следующее:
По сравнению с другими подходами, наши модели не только интегрируют информацию из отдельных эталонных геномов, но и используют последовательности ДНК из более чем 3200 различных геномов человека, а также 850 геномов широкого спектра видов, включая модельные и немодельные организмы. Благодаря надежной и обширной оценке мы показываем, что эти большие модели обеспечивают чрезвычайно точное предсказание молекулярного фенотипа по сравнению с существующими методами.
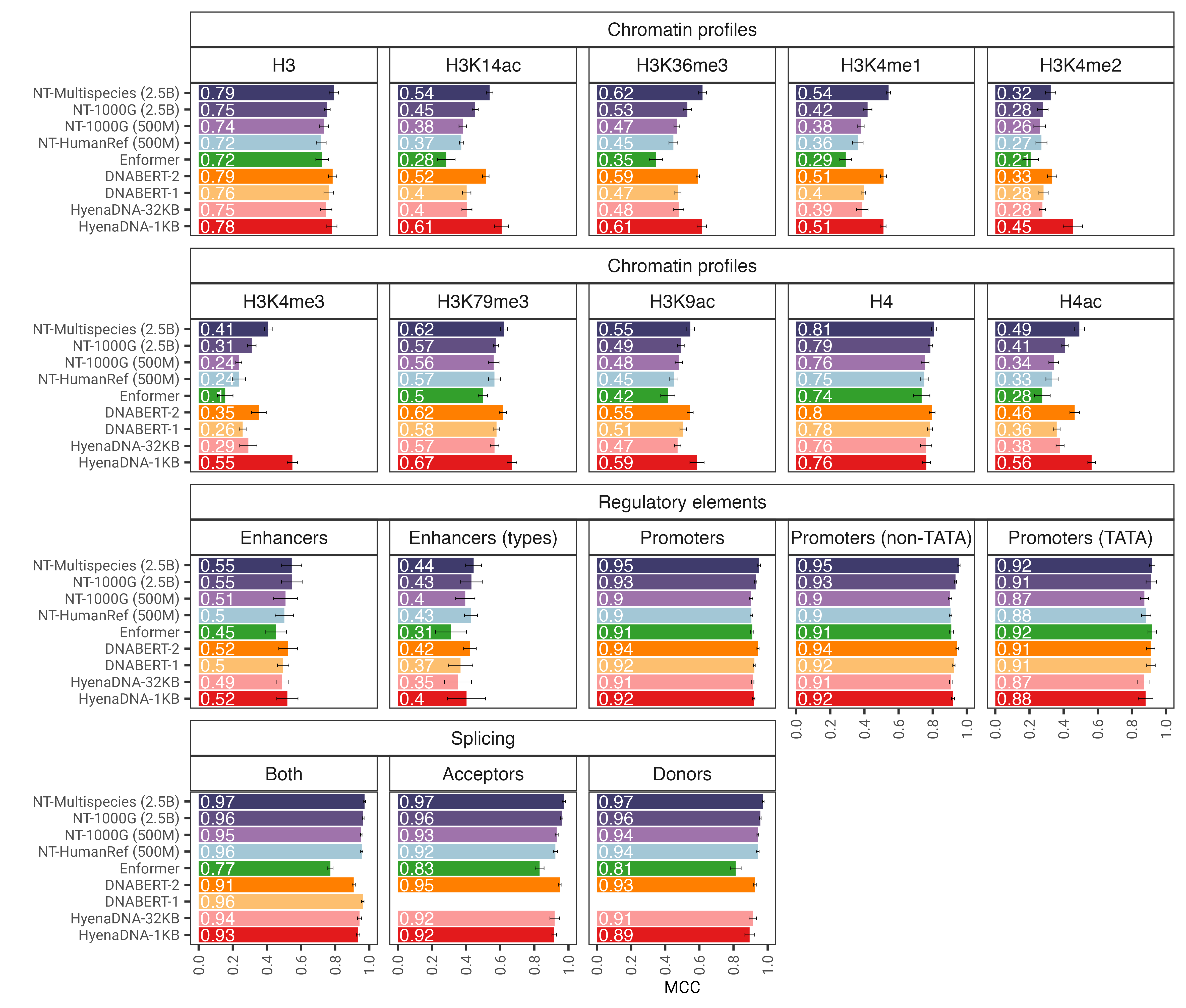
Рис. 1. Модель nucleotide transformer после точной настройки точно предсказывает различные задачи геномики. Мы показываем результаты производительности для последующих задач для точно настроенных моделей трансформаторов. Столбики ошибок представляют собой 2 стандартных отклонения, полученные в результате 10-кратной перекрестной проверки.
В этой работе мы представляем новую основополагающую модель большого языка, обученную на эталонных геномах 48 видов растений с преимущественным акцентом на виды сельскохозяйственных культур. Мы оценили производительность AgroNT по нескольким задачам прогнозирования, начиная от регуляторных функций, обработки РНК и экспрессии генов, и показали, что AgroNT может достичь самых современных показателей.
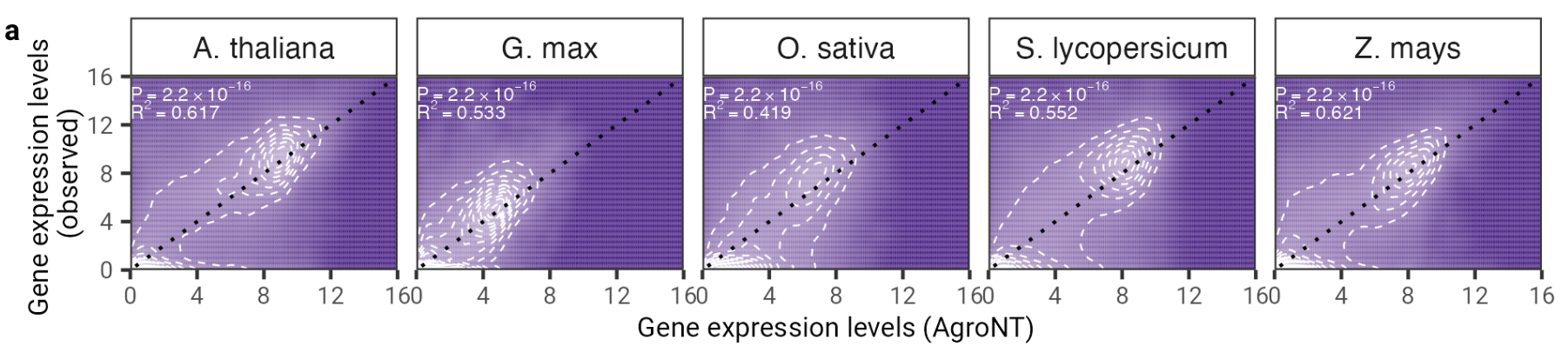
Рис. 2: AgroNT обеспечивает прогнозирование экспрессии генов у разных видов растений. Прогнозирование экспрессии генов-удерживаемых генов во всех тканях коррелирует с наблюдаемыми уровнями экспрессии генов. Показаны коэффициент детерминации (R 2 ) из линейной модели и связанные с ним значения P между прогнозируемыми и наблюдаемыми значениями.
Чтобы использовать код и предварительно обученные модели, просто:
pip install . .Затем вы можете загрузить и сделать вывод с помощью любой из наших девяти моделей, используя всего несколько строк кода:
import haiku as hk
import jax
import jax . numpy as jnp
from nucleotide_transformer . pretrained import get_pretrained_model
# Get pretrained model
parameters , forward_fn , tokenizer , config = get_pretrained_model (
model_name = "500M_human_ref" ,
embeddings_layers_to_save = ( 20 ,),
max_positions = 32 ,
)
forward_fn = hk . transform ( forward_fn )
# Get data and tokenize it
sequences = [ "ATTCCGATTCCGATTCCG" , "ATTTCTCTCTCTCTCTGAGATCGATCGATCGAT" ]
tokens_ids = [ b [ 1 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens_str = [ b [ 0 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens = jnp . asarray ( tokens_ids , dtype = jnp . int32 )
# Initialize random key
random_key = jax . random . PRNGKey ( 0 )
# Infer
outs = forward_fn . apply ( parameters , random_key , tokens )
# Get embeddings at layer 20
print ( outs [ "embeddings_20" ]. shape )Поддерживаемые названия моделей:
Вы также можете запустить наши модели и найти дополнительные примеры кода в Google Colab.
Благодаря Jax код работает как на графическом процессоре, так и на TPU!
Наша вторая версия модели nucleotide transformer v2 включает ряд архитектурных изменений, которые оказались более эффективными: вместо использования изученных позиционных вложений мы используем вращающиеся вложения, которые используются на каждом уровне внимания, и закрытые линейные блоки со взмахом активации без смещения. Эти улучшенные модели также принимают последовательности длиной до 2048 токенов, что приводит к увеличению контекстного окна до 12 Кбит/с. Вдохновленные законами масштабирования Chinchilla, мы также обучили наши модели NT-v2 на нашем многовидовом наборе данных для более длительного периода (токены 300B для моделей 50M и 100M; токены 1T для модели 250M и 500M) по сравнению с моделями v1 (токены 300B). для всех четырех моделей).
Слои преобразователя имеют 1-индексный индекс, что означает, что вызов get_pretrained_model с аргументами model_name="500M_human_ref" и embeddings_layers_to_save=(1, 20,) приведет к извлечению внедрений после первого и 20-го слоев преобразователя. Для трансформаторов, использующих головку Roberta LM, общепринятой практикой является извлечение окончательных вложений после нормы первого слоя головки LM, а не после последнего блока трансформатора. Следовательно, если get_pretrained_model вызывается со следующими аргументами embeddings_layers_to_save=(24,) , внедрения не будут извлечены после финального слоя преобразователя, а скорее после нормы первого слоя головы LM.
Модели SegmentNT используют nucleotide transformer (NT), из которого мы удалили голову языковой модели и заменили ее одномерной головкой сегментации U-Net, чтобы предсказать расположение нескольких типов элементов геномики в последовательности с разрешением в один нуклеотид. Мы представляем два разных варианта модели для 14 различных классов элементов геномики человека во входных последовательностях размером до 30 КБ. К ним относятся генные (гены, кодирующие белки, днРНК, 5'UTR, 3'UTR, экзон, интрон, акцепторные и донорные сайты сплайсинга) и регуляторные (сигнал полиА, тканеинвариантные и тканеспецифические промоторы и энхансеры, а также CTCF-связанные сайты) элементы. SegmentNT достигает превосходной производительности по сравнению с современной архитектурой сегментации U-Net, используя предварительно обученные веса NT, и демонстрирует нулевое обобщение до 50 Кбит/с.
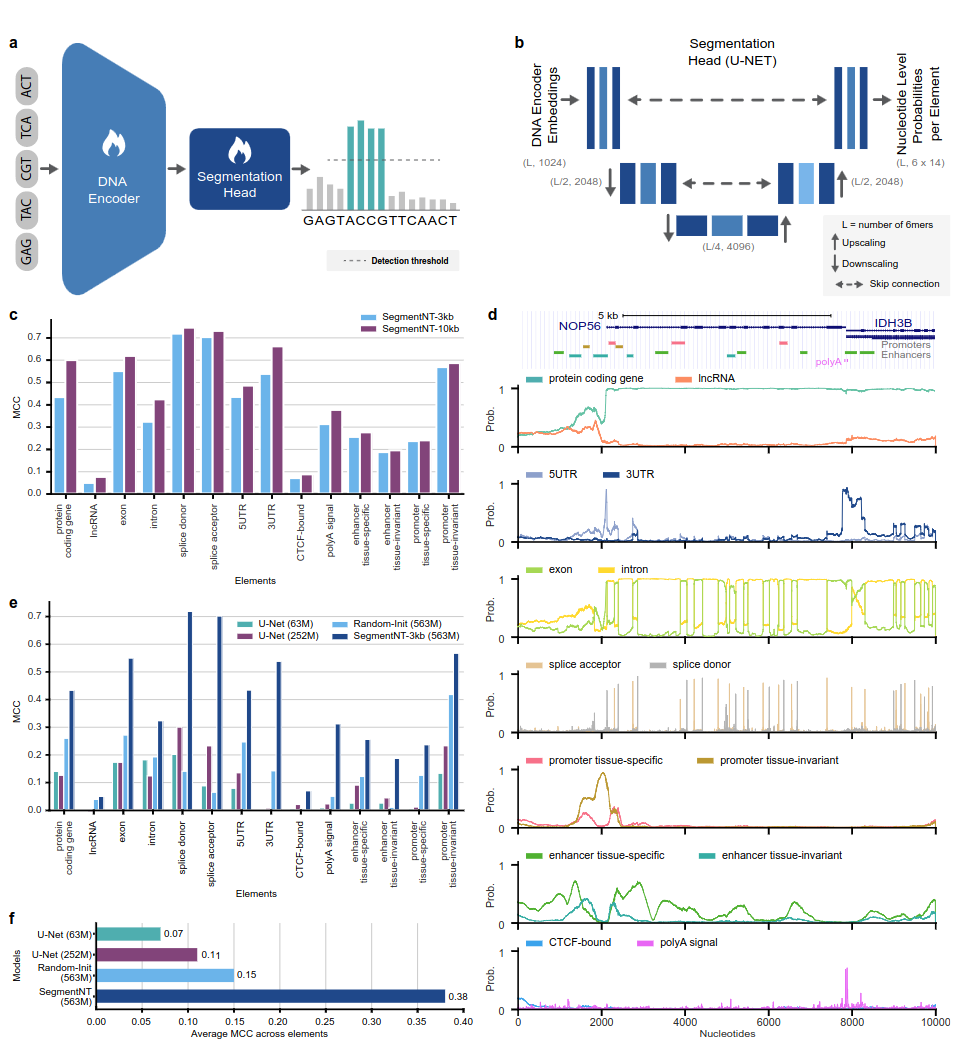
Рис. 1: SegmentNT локализует элементы геномики с разрешением нуклеотидов.
Чтобы использовать код и предварительно обученные модели, просто:
pip install . .Затем вы можете скачать и вывести последовательность с любой из наших моделей, используя всего несколько строк кода:
rescaling factor установлен на тот, который использовался во время обучения. Если вам нужно сделать вывод о последовательностях между 30 и 50 кбит/с, обязательно передайте аргумент rescaling_factor в функцию get_pretrained_segment_nt_model со значением rescaling_factor = max_num_nucleotides / max_num_tokens_nt , где num_dna_tokens_inference — это количество токенов при выводе (т. е. 6669 для последовательности 40008 пар оснований), а max_num_tokens_nt — максимальное количество токенов, на которых был обучен основной нуклеотид-трансформатор, т.е. 2048 .
? В блокноте examples/inference_segment_nt.ipynb показано, как сделать вывод по последовательности размером 50 КБ и построить график вероятностей для воспроизведения рисунка 3 статьи.
? Модели SegmentNT не обрабатывают никакие «N» во входной последовательности, поскольку каждый нуклеотид должен быть токенизирован как 6-мер, чего не может быть при использовании последовательностей, содержащих одну или несколько пар оснований «N».
import haiku as hk
import jax
import jax . numpy as jnp
from nucleotide_transformer . pretrained import get_pretrained_segment_nt_model
# Initialize CPU as default JAX device. This makes the code robust to memory leakage on
# the devices.
jax . config . update ( "jax_platform_name" , "cpu" )
backend = "cpu"
devices = jax . devices ( backend )
num_devices = len ( devices )
print ( f"Devices found: { devices } " )
# The number of DNA tokens (excluding the CLS token prepended) needs to be dividible by
# 2 to the power of the number of downsampling block, i.e 4.
max_num_nucleotides = 8
assert max_num_nucleotides % 4 == 0 , (
"The number of DNA tokens (excluding the CLS token prepended) needs to be dividible by"
"2 to the power of the number of downsampling block, i.e 4." )
parameters , forward_fn , tokenizer , config = get_pretrained_segment_nt_model (
model_name = "segment_nt" ,
embeddings_layers_to_save = ( 29 ,),
attention_maps_to_save = (( 1 , 4 ), ( 7 , 10 )),
max_positions = max_num_nucleotides + 1 ,
)
forward_fn = hk . transform ( forward_fn )
apply_fn = jax . pmap ( forward_fn . apply , devices = devices , donate_argnums = ( 0 ,))
# Get data and tokenize it
sequences = [ "ATTCCGATTCCGATTCCAACGGATTATTCCGATTAACCGATTCCAATT" , "ATTTCTCTCTCTCTCTGAGATCGATGATTTCTCTCTCATCGAACTATG" ]
tokens_ids = [ b [ 1 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens_str = [ b [ 0 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens = jnp . asarray ( tokens_ids , dtype = jnp . int32 )
random_key = jax . random . PRNGKey ( seed = 0 )
keys = jax . device_put_replicated ( random_key , devices = devices )
parameters = jax . device_put_replicated ( parameters , devices = devices )
tokens = jax . device_put_replicated ( tokens , devices = devices )
# Infer on the sequence
outs = apply_fn ( parameters , keys , tokens )
# Obtain the logits over the genomic features
logits = outs [ "logits" ]
# Transform them in probabilities
probabilities = jnp . asarray ( jax . nn . softmax ( logits , axis = - 1 ))[..., - 1 ]
print ( f"Probabilities shape: { probabilities . shape } " )
print ( f"Features inferred: { config . features } " )
# Get probabilities associated with intron
idx_intron = config . features . index ( "intron" )
probabilities_intron = probabilities [..., idx_intron ]
print ( f"Intron probabilities shape: { probabilities_intron . shape } " )Поддерживаемые названия моделей:
Благодаря Jax код работает как на графическом процессоре, так и на TPU!
Модели обучаются на последовательностях длиной до 1000 токенов, включая токен <CLS>, автоматически добавляемый в начало последовательности. Токенизатор начинает токенизацию слева направо, группируя буквы «A», «C», «G» и «T» в 6-меры. Буква «N» выбрана так, чтобы не группироваться внутри k-меров, поэтому всякий раз, когда токенизатор встречает букву «N» или если количество нуклеотидов в последовательности не кратно 6, он токенизирует нуклеотиды без группировки. их. Примеры приведены ниже:
dna_sequence_1 = "ACGTGTACGTGCACGGACGACTAGTCAGCA"
tokenized_dna_sequence_1 = [ < CLS > , < ACGTGT > , < ACGTGC > , < ACGGAC > , < GACTAG > , < TCAGCA > ]
dna_sequence_2 = "ACGTGTACNTGCACGGANCGACTAGTCTGA"
tokenized_dna_sequence_2 = [ < CLS > , < ACGTGT > , < A > , < C > , < N > , < TGCACG > , < G > , < A > , < N > , < CGACTA > , < GTCTGA > ]Таким образом, все преобразователи v1 и v2 могут принимать последовательности длиной до 5994 и 12282 нуклеотидов соответственно, если внутри нет буквы «N».
Коллекция моделей, представленная в этом репозитории, доступна в пространствах Huggingface Instadeep здесь: Пространство nucleotide transformer и Пространство агронуклеотидного nucleotide transformer !
Мы благодарим Машу Роллер, а также сотрудников Ростлаба, особенно Тобиаса Олени, Ивана Колударова и Бурхарда Роста, за конструктивные дискуссии, которые помогли определить интересные направления исследований. Кроме того, мы выражаем благодарность всем тем, кто размещает экспериментальные данные в общедоступных базах данных, тем, кто поддерживает эти базы данных, и тем, кто делает аналитические и прогнозные методы бесплатными. Мы также благодарим команду разработчиков Jax.
Если вы найдете этот репозиторий полезным в своей работе, добавьте соответствующую ссылку на любую из наших связанных статей:
Бумага nucleotide transformer :
@article { dalla2023nucleotide ,
title = { The nucleotide transformer : Building and Evaluating Robust Foundation Models for Human Genomics } ,
author = { Dalla-Torre, Hugo and Gonzalez, Liam and Mendoza Revilla, Javier and Lopez Carranza, Nicolas and Henryk Grywaczewski, Adam and Oteri, Francesco and Dallago, Christian and Trop, Evan and Sirelkhatim, Hassan and Richard, Guillaume and others } ,
journal = { bioRxiv } ,
pages = { 2023--01 } ,
year = { 2023 } ,
publisher = { Cold Spring Harbor Laboratory }
}Агронуклеотидная бумага nucleotide transformer :
@article { mendoza2024foundational ,
title = { A foundational large language model for edible plant genomes } ,
author = { Mendoza-Revilla, Javier and Trop, Evan and Gonzalez, Liam and Roller, Ma{v{s}}a and Dalla-Torre, Hugo and de Almeida, Bernardo P and Richard, Guillaume and Caton, Jonathan and Lopez Carranza, Nicolas and Skwark, Marcin and others } ,
journal = { Communications Biology } ,
volume = { 7 } ,
number = { 1 } ,
pages = { 835 } ,
year = { 2024 } ,
publisher = { Nature Publishing Group UK London }
}Сегментная бумага NT
@article { de2024segmentnt ,
title = { SegmentNT: annotating the genome at single-nucleotide resolution with DNA foundation models } ,
author = { de Almeida, Bernardo P and Dalla-Torre, Hugo and Richard, Guillaume and Blum, Christopher and Hexemer, Lorenz and Gelard, Maxence and Pandey, Priyanka and Laurent, Stefan and Laterre, Alexandre and Lang, Maren and others } ,
journal = { bioRxiv } ,
pages = { 2024--03 } ,
year = { 2024 } ,
publisher = { Cold Spring Harbor Laboratory }
}Если у вас есть какие-либо вопросы или отзывы о коде и моделях, пожалуйста, свяжитесь с нами.
Благодарим Вас за интерес к нашей работе!


