ClockstaR
1.0.0

سيباستيان دوشين، مارتينا مولاك، وسيمون واي دبليو هو.
مختبر البيئة الجزيئية والتطور وعلم الوراثة (MEEP).
كلية العلوم البيولوجية
جامعة سيدني
10 يونيو 2015
partition_data_partitionfinder('drag fasta file with concatenated data here', 'drag partition finder output here')
optim.trees.interactive(folder.parts = 'path to your folder with fasta files and tree topology here')
تنفيذ تحسين مسافات الأشجار باستخدام مشتق مسافة BSD
تنفيذ نسخة متوازية لمسافة الطوبولوجيا
كتابة برنامج تعليمي عن التجميع عن بعد طوبولوجيا
دمج مولد النماذج لاختبار النموذج
قم بدمج RaxML لتحقيق أقصى قدر من التحسين لأطوال الفروع والطوبولوجيا
يعد تقدير الجداول الزمنية التطورية باستخدام مجموعات بيانات متعددة الجينات ممارسة شائعة في دراسات النشوء والتطور. يمكن تقسيم مجموعات البيانات متعددة الجينات حسب الجين أو موضع الكودون أو كليهما. في هذا البرنامج التعليمي، نشير إلى "مجموعات البيانات الفرعية" باعتبارها جينات فردية أو أي وحدة فرعية من مجموعة البيانات متعددة الجينات. يشير مصطلح "الأقسام" إلى مجموعة من مجموعات البيانات الفرعية.
على الرغم من أنه يمكن تسلسل مجموعات البيانات الفرعية وتحليلها باستخدام نموذج ساعة مريح واحد، إلا أن أنماط تباين معدل النسب يمكن أن تختلف بين مجموعات البيانات الفرعية حتى عندما تكون طبولوجيا الشجرة الخاصة بها متطابقة. على سبيل المثال، يمكن أن يختلف تباين معدل النسب في جينات الميتوكوندريا عن تباين الجينات النووية. ولذلك، يمكن تعيين نماذج مختلفة للساعة المريحة لمجموعات فرعية مختلفة من البيانات من أجل تحسين تقديرات الجداول الزمنية التطورية والملاءمة الإحصائية (انظر Duchene and Ho., 2014a).
هناك عدد كبير من الطرق التي يمكن من خلالها تقسيم مجموعات البيانات متعددة الجينات. من الأساليب الشائعة لمقارنة مخططات التقسيم استخدام عوامل بايز أو المعايير القائمة على الاحتمالية لملاءمة النموذج. ومع ذلك، في معظم الحالات، من غير الممكن اختبار جميع مخططات التقسيم الممكنة، خاصة مع الطرق المكثفة حسابيًا لحساب عوامل بايز.
تقدر ClockstaR أطوال فرع النشوء والتطور لكل مجموعة فرعية من البيانات. يتم حساب مسافة درجة الفرع، المعروفة باسم sBSDmin، لكل زوج من الأشجار كمقياس للاختلاف في أنماط التباين بين معدلات النسب. تُستخدم هذه المسافات لاستنتاج أفضل استراتيجية تقسيم باستخدام إحصائيات GAP مع خوارزمية تجميع PAM، كما تم تنفيذها في مجموعة الحزم (Maechler et al., 2012) (لمزيد من التفاصيل حول مقياس sBSDmin، راجع Duchene et al., 2014b) .
ClockstaR عبارة عن حزمة R لتحليلات الساعة الجزيئية التطورية لمجموعات البيانات متعددة الجينات. ويستخدم أنماط التباين بين معدل النسب للجينات المختلفة لتحديد استراتيجية تقسيم الساعة. تستخدم الطريقة مقياس مسافة شجرة النشوء والتطور وخوارزمية التعلم الآلي غير الخاضعة للرقابة لتحديد العدد الأمثل لأجزاء الساعة، وأي الجينات يجب تحليلها تحت كل جزء من الأجزاء. يمكن استخدام استراتيجية التقسيم المحددة في ClocsktaR لتحليل الساعة الجزيئية اللاحقة باستخدام برامج مثل BEAST وMrBayes وPhyloBayes وغيرها.
يرجى اتباع هذا الرابط للنشر الأصلي.
يتطلب ClockstaR تثبيت R. كما يتطلب أيضًا بعض تبعيات R، والتي يمكن الحصول عليها من خلال R، كما هو موضح أدناه.
يرجى إرسال أي طلبات أو أسئلة إلى سيباستيان دوشين (sebastian.duchene[at]sydney.edu.au). يمكن العثور على بعض البرامج والموارد الأخرى في مختبر البيئة الجزيئية والتطور وعلم الوراثة بجامعة سيدني.
قم بتنزيل هذا المستودع كملف مضغوط وقم بفك ضغطه. تستخدم التعليمات التالية مجلد Clockstar_example_data، الذي يحتوي على بعض ملفات fasta وشجرة النشوء والتطور بتنسيق newick. افتح أيًا من هذه الملفات في محرر نصوص، مثل text wrangler. تمت محاكاة هذه البيانات تحت أربعة أنماط من تباين معدل التطور. لاحظ أن الشجرة هي طوبولوجيا الشجرة لجميع الجينات أو أقسام البيانات. لتشغيل ClockstaR، يرجى تنسيق بياناتك بشكل مشابه لبيانات المثال في Clockstar_example_data.
يمكن تثبيت ClockstaR مباشرة من GitHub. وهذا يتطلب حزمة devtools. اكتب الكود التالي في موجه R لتثبيت جميع الأدوات الضرورية (لاحظ أنك ستحتاج إلى اتصال بالإنترنت لتنزيل الحزم مباشرة):.
install . packages ( " devtools " )
library (devtools)
install_github ( ' ClockstaR ' , ' sebastianduchene ' )بعد التنزيل والتثبيت، قم بتحميل ClockstaR بمكتبة الوظائف.
library (ClockstaR2)لمشاهدة مثال على كيفية تشغيل البرنامج اكتب:
example (ClockstaR2)يستخدم باقي هذا البرنامج التعليمي مجلد Clockstar_example_data
الخطوة الأولى هي الحصول على أشجار الجينات لكل من المحاذاة. للقيام بذلك، نستخدم طوبولوجيا الشجرة، ونحسن أطوال الفروع باستخدام كل من محاذاة الجينات الفردية، في هذه الحالة من A1.fasta إلى C3.fasta. إذا كان لديك أشجار الجينات، فاحفظها بتنسيق newick في ملف وانتقل إلى الخطوة التالية (تشغيل Clockstar بشكل تفاعلي).
اكتب الكود التالي في موجه R واضغط على Enter:
optim . trees . interactive ()إذا تلقيت رسالة خطأ حول تثبيت الحزمة phangorn، يرجى استخدام هذا الرمز ثم تكرار optim.trees.interactive()
install . packcages ( " phangorn " )ستقوم ClockstaR بطباعة الرسالة التالية:
Please drag a folder with the data subsets and a tree topology . The files should be in FASTA format, and the trees in NEWICKاسحب مجلد Clockstar_example_data إلى وحدة التحكم R واكتب إدخال. لاحظ أن المجلد يجب أن يحتوي فقط على التحالفات بتنسيق FASTA، وطوبولوجيا الشجرة بتنسيق NEWICK. ستظهر لك الرسالة التالية:
What should be the name of the file to save the optimised trees ?اكتب اسم الملف للأشجار الأمثل. في هذه الحالة سوف نستخدم "example.trees"
example . treesعند هذه النقطة، سوف تسأل ClockstaR عما إذا كان ينبغي استخدام نموذج بديل منفصل لكل جين، أو استخدام JC في جميع الحالات. وبما أنه تمت محاكاة هذه البيانات تحت JC، فسنكتب "n" ونضغط على زر الإدخال. اكتب "y" لتحديد كل نموذج بديل على حدة.
بعد كتابة "n" والضغط على زر الإدخال، سيبدأ تشغيل ClockstaR. سيقوم بطباعة أشجار الجينات في جهاز الرسومات. إذا تم تجذير الشجرة المحددة، فقد تطبع أيضًا بعض التحذيرات، والتي يمكن تجاهلها بأمان.
افتح مجلد Clockstar_example_data. سوف تجد ملفًا بالاسم "example.trees"، كما هو محدد في بضع خطوات أعلاه. افتح example.trees في محرر النصوص. ويحتوي على كل شجرة جينات وأسماء الأشجار، وفقًا لأسماء محاذاة الجينات. يجب أن يبدو مثل هذا:
A1 . fasta (( t1 : 0.01504695462 ,( t2 : 0.00987 ...
A2 . fasta (( t1 : 0.01520523401 ,( t2 : 0.01317 ...
A3 . fasta (( t1 : 0.01519309467 ,( t2 : 0.01092 ...
.
.
.سيتم استخدام هذا الملف الذي يحتوي على الأشجار في الخطوة التالية.
في هذه الخطوة، من الضروري وجود أشجار الجينات في ملف، مثل ذلك الذي تم الحصول عليه في الخطوة السابقة.
افتح R وقم بتحميل ClockstaR كما هو موضح أعلاه. اكتب الكود التالي في الموجه:
clockstar . interactive ()ستقوم ClockstaR بطباعة الرسالة التالية:
please drag or type in the path to your gene trees file in NEWICK format :اسحب الملف الذي يحتوي على أشجار الجينات إلى وحدة التحكم R. إذا اتبعت الخطوة السابقة، فسيُسمى الملف example.trees. اكتب أدخل.
اعتمادًا على الحزم التي قمت بتثبيتها، قد يسألك ClockstaR عما إذا كان يجب تشغيله بالتوازي. وهذا فعال لمجموعات البيانات الكبيرة. لكن بالنسبة لبيانات المثال، فلن يحدث ذلك فرقًا كبيرًا، لذا اكتب "n" إذا رأيت هذه الرسالة ثم اكتب إدخال:
Packages foreach and doParallel are available for parallel computation
Should we run ClockstaR in parallel (y / n) ? (This is good for large data sets)سيبدأ تشغيل Clockstar الآن. يجب أن يبدو الإخراج على الشاشة كما يلي:
[ 1 ] " Calculating sBSDmin distances between all pairs of trees "
[ 1 ] " Estimating tree distances "
[ 1 ] " estimating distances 1 of 11 "
[ 1 ] " estimating distances 2 of 11 "
[ 1 ] " estimating distances 3 of 11 "
[ 1 ] " estimating distances 4 of 11 "
[ 1 ] " estimating distances 5 of 11 "
.
.
.بعد تقدير مسافات الشجرة (الموصوفة في المنشور الأصلي)، ستقوم ClockstaR بطباعة الرسالة التالية:
" I finished calculating the sBSDmin distances between trees "
The settings for clustering with ClockstaR are :
PAM clustering algorithm
K from 1 to number of data subsets - 1
SEmax criterion to select the optimal k
500 bootstrap replicates
Are these correct ? (y / n)هذه هي إعدادات خوارزمية التجميع. وهي مناسبة لمعظم مجموعات البيانات، لذا في هذا المثال يمكننا كتابة "y" ثم إدخالها. وبكتابة "n" يمكننا تغيير هذه الإعدادات، لمزيد من التفاصيل، راجع Kaufman and Rousseeuw (2009).
سيقوم ClockstaR الآن بتشغيل خوارزمية التجميع. في النهاية سيتم طباعة أفضل عدد من الأقسام ويسأل ما إذا كان ينبغي حفظ النتائج في ملف pdf:
[ 1 ] " ClockstaR has finished running "
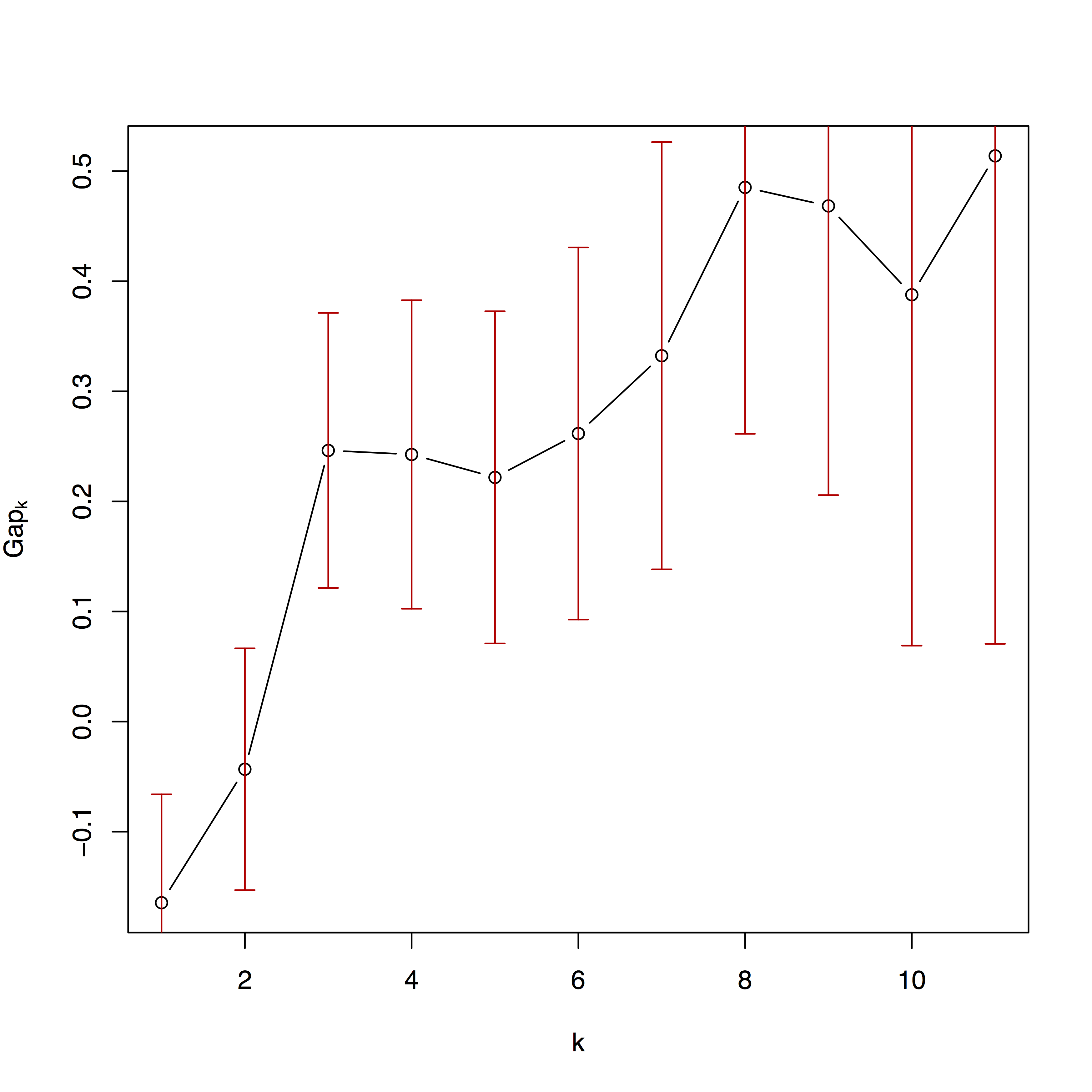
[ 1 ] " The best number of partitions for your data set is: 3 "
Do you wish to save the results in a pdf file ? (y / n)اكتب "y" ثم أدخل.
سيطلب ClockstaR بعد ذلك اسم ملفات الإخراج:
What should be the name and path of the output file ?في هذا المثال، اكتب "example_run" وأدخل، ولكن يمكن استخدام أي اسم.
الآن افتح مجلد Clockstar_example_data وافتح ملفي pdf، example_run_gapstats.pdf وexample_run_matrix.pdf.
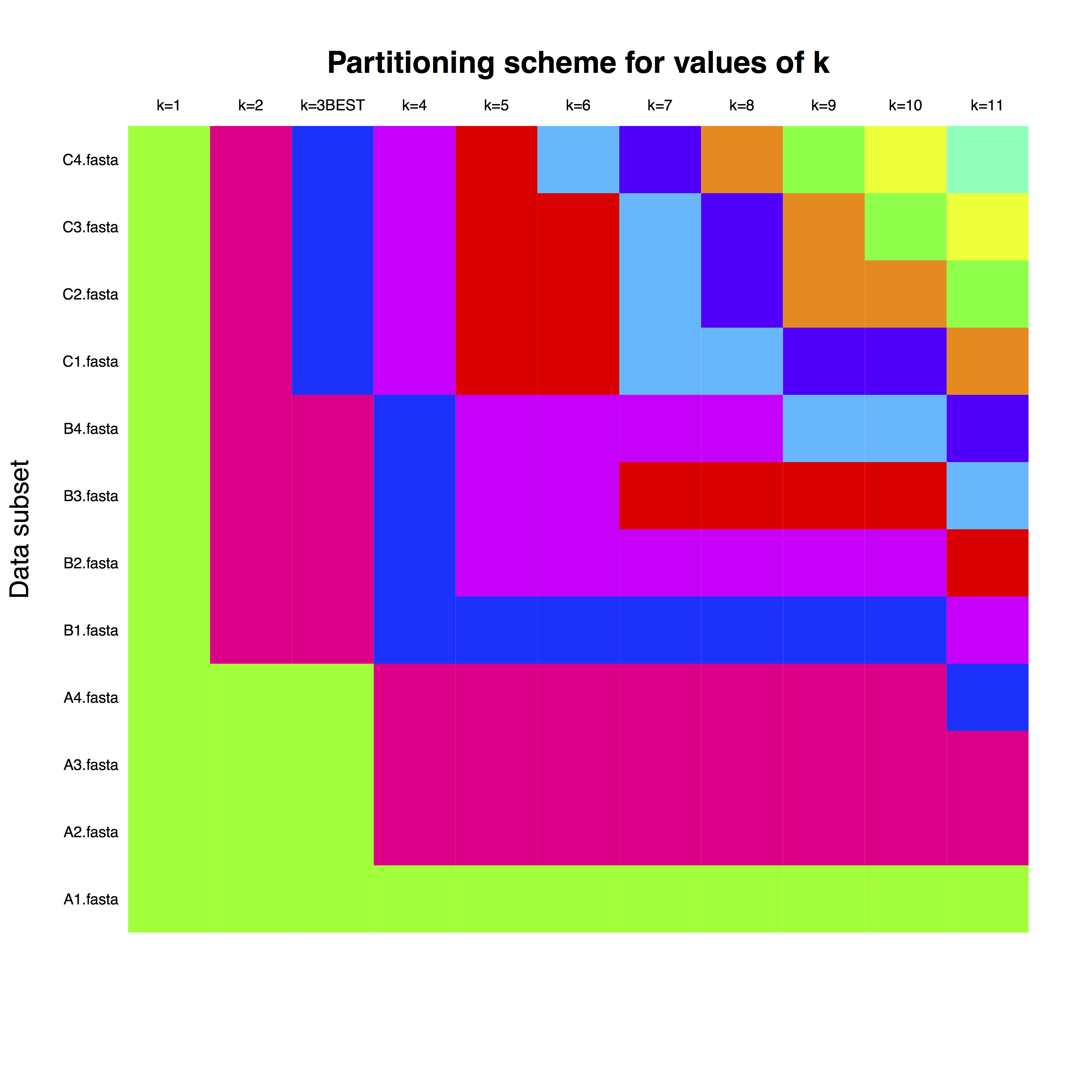
example_run_matrix عبارة عن مصفوفة، حيث تتوافق الصفوف مع كل جين، كما هو مذكور في ملفات FASTA. تمثل الأعمدة عدد الأقسام، وتمثل الألوان تخصيص كل جين لقسم الساعة. على سبيل المثال، بالنسبة لـ k = 3، وهو أفضل عدد من الأقسام، يمكن للمرء استخدام أقسام ساعة منفصلة للجينات ذات الحروف A وB وC.
المؤامرة الثانية هي ملاءمة خوارزميات التجميع عبر أعداد مختلفة من الأقسام. تتوفر المزيد من التفاصيل في كوفمان وروسيو (2009) وفي الوثائق الخاصة بمجموعة الحزم.
يمكن تشغيل ClockstaR باستخدام إعدادات مخصصة أخرى. يرجى الاطلاع على الوثائق للحصول على تفاصيل أخرى أو مراسلتي إذا كانت لديك أي أسئلة على sebastian.duchene[at]sydney.edy.au.
تم تصميم الشعار بواسطة Jun Tong
دوشين، س.، وهو، سي واي (2014أ). استخدام نماذج متعددة للساعة المريحة لتقدير الجداول الزمنية التطورية من بيانات تسلسل الحمض النووي. علم الوراثة الجزيئي والتطور (77): 65-70.
دوشين، إس، مولاك، إم، وهو، إس واي (2014ب). ClockstaR: اختيار عدد نماذج الساعة المريحة في تحليل التطور الجزيئي. المعلوماتية الحيوية 30 (7): 1017-1019.
كوفمان، إل.، وروسيو، بي جي (2009). العثور على مجموعات في البيانات: مقدمة للتحليل العنقودي (المجلد 344). جون وايلي وأولاده.


