reference_database_creator
bug fix --in-silico-pcr --untrimmed
سرطان البحر ( ج إعادة ر قواعد البيانات المرجعية ل أ mplicon- ب ased س التسلسل) هو برنامج متعدد الاستخدامات يقوم بإنشاء قواعد بيانات مرجعية منسقة للتحليل الميتاجينومي. يتكون سير عمل CRABS من سبع وحدات: (1) تنزيل البيانات من المستودعات عبر الإنترنت؛ (2) استيراد البيانات التي تم تنزيلها إلى تنسيق CRABS؛ (ثالثا) استخراج مناطق amplicon من خلال تحليل PCR silico ؛ (رابعا) استرداد الأمبليكونات دون مناطق الربط التمهيدي من خلال المحاذاة مع الباركود المستخرج من السيليكو ؛ (v) تنظيم قاعدة البيانات المحلية وتعيينها فرعيًا عبر معلمات تصفية متعددة؛ (6) تصدير قاعدة البيانات المحلية بتنسيقات مختلفة وفقًا لمتطلبات المصنف التصنيفي؛ و(6) وظائف ما بعد المعالجة، أي التصورات، لاستكشاف قاعدة البيانات المرجعية المحلية وتقديم نظرة عامة موجزة عنها. وتنقسم هذه الوحدات السبع إلى ثمانية عشر وظيفة، وهي موضحة أدناه. بالإضافة إلى ذلك، يتم توفير رمز المثال لكل وظيفة من الوظائف الثمانية عشر. أخيرًا، يتم توفير برنامج تعليمي لإنشاء قاعدة بيانات مرجعية لأسماك القرش المحلية لمجموعة MiFish-E التمهيدية في نهاية مستند README هذا لتوفير مثال لبرنامج نصي كمرجع.
يسعدنا أن نعلن أن CRABS قد شهد تحديثًا كبيرًا وإعادة تصميم التعليمات البرمجية بناءً على تعليقات المستخدمين، والتي نأمل أن تؤدي إلى تحسين تجربة المستخدم في بناء قاعدة البيانات المرجعية المحلية الخاصة بك!
تجدون أدناه قائمة بالميزات والتحسينات المضافة إلى CRABS v 1.0.0 :
يمكن الآن تنزيل CRABS v 1.0.0 يدويًا عن طريق استنساخ مستودع GitHub هذا (راجع 4.1 التثبيت اليدوي للحصول على معلومات مفصلة). سنقوم بتحديث حاوية Docker وحزمة conda في أقرب وقت ممكن لتسهيل التثبيت السهل للإصدار الأحدث.
عند استخدام CRABS في مشاريعك البحثية، يرجى الاستشهاد بالمقالة التالية:
[Jeunen, G.-J., Dowle, E., Edgecombe, J., von Ammon, U., Gemmell, N. J., & Cross, H. (2022). crabs—A software program to generate curated reference databases for metabarcoding sequencing data. Molecular Ecology Resources, 00, 1– 14.](https://doi.org/10.1111/1755-0998.13741)
CRABS عبارة عن مجموعة أدوات لسطر الأوامر فقط تعمل على بيئات Unix/Linux النموذجية وهي مكتوبة حصريًا بلغة python3. ومع ذلك، يستخدم CRABS وحدة المعالجة الفرعية في python لتشغيل عدة أوامر في بناء جملة bash للتحايل على الخصوصيات الخاصة بـ python وزيادة سرعة التنفيذ. نحن نقدم ثلاث طرق لتثبيت CRABS. للحصول على أحدث إصدار من CRABS، نوصي بالتثبيت اليدوي عن طريق استنساخ مستودع GitHub هذا وتثبيت 10 تبعيات بشكل منفصل (تعليمات التثبيت لجميع التبعيات متوفرة في 4.1 التثبيت اليدوي). يمكن أيضًا تثبيت CRABS عبر Docker وconda. تسمح كلتا الطريقتين بالتثبيت السهل عن طريق التثبيت المشترك لجميع التبعيات تلقائيًا. نحن نهدف إلى الحفاظ على تحديث حاوية Docker وحزمة conda، على الرغم من إمكانية حدوث تأخير معين في التحديث إلى الإصدار الأحدث، خاصة بالنسبة لحزمة conda. وفيما يلي تفاصيل لجميع الأساليب الثلاثة.
للتثبيت اليدوي، قم أولاً باستنساخ مستودع CRABS. تتطلب هذه الخطوة أن يكون GitHub متاحًا لسطر الأوامر (تعليمات التثبيت لـ GitHub).
git clone https://github.com/gjeunen/reference_database_creator.git
اعتمادًا على إعداداتك، قد يلزم جعل CRABS قابلاً للتنفيذ على نظامك. يمكن تحقيق ذلك باستخدام الكود أدناه.
chmod +x reference_database_creator/crabs
بمجرد تثبيت CRABS، نحتاج إلى التأكد من تثبيت جميع التبعيات وإمكانية الوصول إليها عالميًا. يعمل أحدث إصدار من CRABS (الإصدار v 1.0.0 ) على Python 3.11.7 (أو أي إصدار متوافق مع 3.11.7) ويعتمد على خمس وحدات Python قد لا تأتي بشكل قياسي مع Python، بالإضافة إلى خمسة برامج خارجية. جميع التبعيات مدرجة أدناه، بالإضافة إلى رابط لتعليمات التثبيت. أرقام الإصدارات المقدمة لكل وحدة وبرنامج هي تلك التي تم تطوير CRABS عليها. على الرغم من إمكانية استخدام الإصدارات المتوافقة لكل منها أيضًا.
وحدات بايثون:
البرامج الخارجية:
بمجرد تثبيت CRABS وجميع التبعيات، يمكن إتاحة الوصول إلى CRABS عبر نظام التشغيل باستخدام الكود أدناه.
export PATH="/path/to/crabs/folder:$PATH"
استبدل /path/to/crabs/folder بالمسار الفعلي لمجلد مستودع GitHub على نظام التشغيل، أي المجلد الذي تم إنشاؤه أثناء أمر git clone أعلاه. ستؤدي إضافة كود export إلى ملف .bash_profile أو .bashrc إلى جعل CRABS متاحًا عالميًا في أي وقت.
Docker هو مشروع مفتوح المصدر يسمح بنشر تطبيقات البرامج داخل "حاويات" معزولة عن جهاز الكمبيوتر الخاص بك ويتم تشغيلها من خلال نظام تشغيل مضيف افتراضي يسمى Docker Engine. الميزة الرئيسية لتشغيل عامل الإرساء على الأجهزة الافتراضية هي أنها تستخدم موارد أقل بكثير. ويعني هذا العزل أنه يمكنك تشغيل حاوية Docker على معظم أنظمة التشغيل، بما في ذلك Mac وWindows وLinux. قد تحتاج إلى إعداد حساب مجاني لاستخدام Docker Desktop. يحتوي هذا الرابط على مقدمة رائعة لأساسيات استخدام Docker. إليك رابط للبدء والتوجه نحو الكون المتعدد Docker.
هناك خطوتان فقط لتشغيل Crabs على جهاز الكمبيوتر الخاص بك. أولاً، قم بتثبيت Docker Desktop على جهاز الكمبيوتر الخاص بك، وهو مجاني لمعظم المستخدمين. فيما يلي التعليمات الخاصة بنظام التشغيل Mac ؛ فيما يلي الإرشادات الخاصة بأجهزة الكمبيوتر التي تعمل بنظام Windows ، وإليك الإرشادات الخاصة بنظام التشغيل Linux (معظم منصات Linux الرئيسية مدعومة). بمجرد تثبيت Docker Desktop وتشغيله (يجب أن يكون تطبيق سطح المكتب قيد التشغيل لتتمكن من استخدام أي أوامر عامل إرساء في سطر الأوامر)، ما عليك سوى "سحب" صورة Crabs الخاصة بنا، وستكون جاهزًا للبدء:
docker pull quay.io/swordfish/crabs:0.1.7
على الرغم من سهولة تثبيت تطبيق عامل الإرساء، إلا أن استخدام هذه التطبيقات قد يكون صعبًا بعض الشيء في البداية. لمساعدتك على البدء، قدمنا بعض الأمثلة على الأوامر باستخدام إصدار عامل الإرساء من Crabs. يمكن العثور على هذه الأمثلة في مجلد docker_intro في هذا الريبو . من هذه الأمثلة، يجب أن تكون قادرًا على البدء في إعداد قاعدة بيانات مرجعية كاملة وتكون جاهزًا للبدء. سنستمر في التوسع في هذه الأمثلة واختبار ذلك في العديد من المواقف المختلفة. يرجى طرح الأسئلة وتقديم التعليقات في علامة التبويب "المشاكل".
لتثبيت حزمة conda، يجب عليك أولاً تثبيت conda. انظر هذا الرابط للحصول على التفاصيل. إذا تم تثبيت conda بالفعل، فمن الممارسات الجيدة تحديث أداة conda باستخدام conda update conda قبل تثبيت CRABS.
بمجرد تثبيت conda، اتبع الخطوات أدناه لتثبيت CRABS وجميع التبعيات. تأكد من إدخال الأوامر بالترتيب الذي تظهر به أدناه.
conda create -n CRABS
conda activate CRABS
conda config --add channels bioconda
conda config --add channels conda-forge
conda install -c bioconda crabs
بمجرد إدخال أمر التثبيت، ستقوم conda بمعالجة الطلب (قد يستغرق ذلك دقيقة أو نحو ذلك)، ثم تعرض جميع الحزم والبرامج التي سيتم تثبيتها، وتطلب تأكيد ذلك. اكتب y لبدء التثبيت. بعد انتهاء هذا، يجب أن يكون CRABS جاهزًا للانطلاق.
لقد اختبرنا هذا التثبيت على أنظمة Mac وLinux. لم نختبر بعد نظام Windows الفرعي لنظام التشغيل Linux (WSL).
استخدم الكود أدناه للتحقق من تثبيت CRABS بنجاح واسحب معلومات المساعدة.
crabs -hتقسم معلومات المساعدة الوظائف الثمانية عشر إلى مجموعات مختلفة، حيث تدرج كل مجموعة الوظيفة في الأعلى والمعلمات المطلوبة والاختيارية في الأسفل.

يحتوي CRABS على سبع وحدات تتضمن ثمانية عشر وظيفة:
الوحدة 1: تنزيل البيانات من المستودعات عبر الإنترنت
--download-taxonomy : تنزيل معلومات تصنيف NCBI؛--download-bold : تنزيل بيانات التسلسل من قاعدة بيانات الباركود للحياة (BOLD)؛--download-embl : تنزيل بيانات التسلسل من أرشيف النوكليوتيدات الأوروبي (ENA؛ EMBL)؛--download-mitofish : تنزيل بيانات التسلسل من قاعدة بيانات MitoFish؛--download-ncbi : تنزيل بيانات التسلسل من المركز الوطني لمعلومات التكنولوجيا الحيوية (NCBI).الوحدة 2: استيراد البيانات التي تم تنزيلها إلى تنسيق CRABS
--import : استيراد التسلسلات التي تم تنزيلها أو الرموز الشريطية المخصصة إلى تنسيق CRABS؛--merge : دمج ملفات مختلفة بتنسيق CRABS في ملف واحد.الوحدة 3: استخراج مناطق الأمبليكون من خلال تحليل سيليكو PCR
--in-silico-pcr : استخراج الأمبليكونات من البيانات التي تم تنزيلها عن طريق تحديد موقع مناطق الربط التمهيدي وإزالتها.الوحدة 4: استرداد الأمبليكونات دون مناطق الربط التمهيدي
--pairwise-global-alignment : استرداد amplicons بدون مناطق ربط أولية عن طريق محاذاة التسلسلات التي تم تنزيلها مع الرموز الشريطية المستخرجة من silico .الوحدة 5: تنظيم قاعدة البيانات المحلية وتعيينها فرعيًا عبر معلمات تصفية متعددة
--dereplicate : تجاهل التسلسلات المكررة؛--filter : تجاهل التسلسلات عبر معلمات تصفية متعددة؛--subset : مجموعة فرعية من قاعدة البيانات المحلية للاحتفاظ أو استبعاد مجموعات تصنيفية محددة.الوحدة 6: تصدير قاعدة البيانات المحلية
--export : تصدير قاعدة البيانات بتنسيق CRABS إلى تنسيقات مختلفة وفقًا لمتطلبات المصنف التصنيفي المراد استخدامه.الوحدة 7: وظائف ما بعد المعالجة لاستكشاف وتقديم نظرة عامة موجزة عن قاعدة البيانات المرجعية المحلية
--diversity-figure : ينشئ مخططًا شريطيًا أفقيًا يعرض عدد الأنواع ومجموعة التسلسلات لكل مستوى محدد مدرج في قاعدة البيانات المرجعية؛--amplicon-length-figure : ينشئ مخططًا خطيًا يصور توزيعات طول الأمبليكون مفصولة بالمجموعة التصنيفية؛--phylogenetic-tree : تنشئ شجرة النشوء والتطور باستخدام الرموز الشريطية من قاعدة البيانات المرجعية لقائمة الأنواع المستهدفة؛--amplification-efficiency-figure : إنشاء رسم بياني شريطي يعرض عدم التطابق في مناطق الربط التمهيدي؛--completeness-table : ينشئ جدول بيانات يحتوي على توفر الرمز الشريطي للمجموعات التصنيفية.يمكن تنزيل بيانات التسلسل الأولية بواسطة CRABS من أربعة مستودعات عبر الإنترنت، بما في ذلك (i) BOLD، (ii) EMBL، (iii) MitoFish، وNCBI. بدءًا من الإصدار v 1.0.0 وما بعده، تم تقسيم تنزيل البيانات من كل مستودع إلى وظيفته الخاصة. بالإضافة إلى ذلك، لا يقوم CRABS بتنسيق البيانات تلقائيًا بعد التنزيل لزيادة المرونة وتمكين تصحيح الأخطاء عند فشل تنزيل البيانات.
إلى جانب تنزيل بيانات التسلسل، يستطيع CRABS أيضًا تنزيل معلومات تصنيف NCBI، والتي يستخدمها CRABS لإنشاء النسب التصنيفية لكل تسلسل.
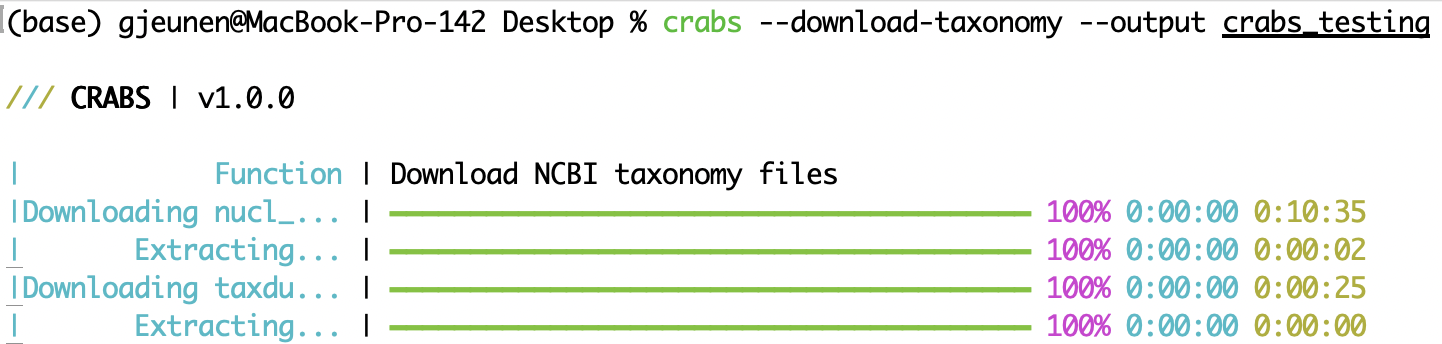
--download-taxonomy لتعيين نسب تصنيفي لكل تسلسل تم تنزيله في قاعدة البيانات المرجعية (انظر 5.2 الوحدة 2)، يجب تنزيل المعلومات التصنيفية. يستخدم CRABS تصنيف NCBI ويقوم بتنزيل ثلاثة ملفات محددة على جهاز الكمبيوتر الخاص بك: (i) ملف يربط أرقام الدخول بالمعرفات التصنيفية ( nucl_gb.accession2taxid )، (ii) ملف يحتوي على معلومات حول اسم التطور المرتبط بكل معرف تصنيفي ( names.dmp ) )، و(3) ملف يحتوي على معلومات حول كيفية ربط المعرفات التصنيفية ( nodes.dmp ). يمكن تحديد دليل الإخراج للملفات التي تم تنزيلها باستخدام المعلمة --output . لاستبعاد إما الملف nucl_gb.accession2taxid أو الملفات names.dmp و nodes.dmp ، يمكن توفير المعلمة --exclude acc2tax أو --exclude taxdump ، على التوالي. لا يقوم الكود الأول أدناه بتنزيل أي ملف، حيث يتم توفير كل من acc2tax و taxdump للمعلمة --exclude . يقوم السطر الثاني من التعليمات البرمجية بتنزيل الملفات الثلاثة جميعها إلى الدليل الفرعي --output crabs_testing . تعرض لقطة الشاشة الموجودة بالأسفل ما تتم طباعته على وحدة التحكم عند تنفيذ هذا السطر من التعليمات البرمجية.
crabs --download-taxonomy --exclude 'acc2taxid,taxdump'
crabs --download-taxonomy --output crabs_testing
--download-bold يتم تنزيل تسلسلات BOLD من خلال موقع BOLD الإلكتروني. يمكن تحديد ملف الإخراج، الذي تم تنظيمه كمستند Fasta المكون من سطرين، باستخدام المعلمة --output . يمكن للمستخدمين تحديد المجموعة التصنيفية التي سيتم تنزيلها باستخدام المعلمة --taxon . نوصي بكتابة حلقة for بسيطة (المثال موضح أدناه) عندما يرغب المستخدمون في تنزيل مجموعات تصنيفية متعددة، وبالتالي الحد من كمية البيانات التي سيتم تنزيلها من BOLD لكل مثيل. ومع ذلك، إذا كان هناك عدد محدود فقط من المجموعات التصنيفية ذات الاهتمام، فيمكن أيضًا فصل أسماء المجموعات التصنيفية بـ | (المثال موضح أدناه). نوصي أيضًا المستخدمين بالتحقق مما إذا كان اسم المجموعة التصنيفية المراد تنزيله مدرجًا في أرشيف BOLD أو ما إذا كانت هناك حاجة لاستخدام أسماء بديلة. على سبيل المثال، لن يؤدي تحديد --taxon Chondrichthyes إلى تنزيل جميع تسلسلات الأسماك الغضروفية من BOLD، نظرًا لأن اسم الفئة هذا غير مدرج في BOLD. يجب على المستخدمين بدلاً من ذلك استخدام --taxon Elasmobranchii في هذه الحالة. يمكن للمستخدمين أيضًا تحديد قصر التنزيل على علامة وراثية معينة من خلال توفير المعلمة --marker . عندما تكون هناك علامات وراثية متعددة ذات أهمية، يجب فصل أسماء العلامات بـ | . علامات ترميز الحمض النووي الرئيسية الأربعة في BOLD هي COI-5P و ITS و matK و rbcL . يعتبر إدخال المعلمة --marker حساسًا لحالة الأحرف.
النهج الموصى به: حلقة for بسيطة لتنزيل البيانات من BOLD لمجموعات تصنيفية متعددة (النهج الموصى به). يقوم الكود الموجود أدناه بتنزيل بيانات Elasmobranchii أولاً، متبوعة بالتسلسلات المخصصة للثدييات. ستتم كتابة البيانات التي تم تنزيلها إلى الدليل الفرعي --output crabs_testing ووضعها في ملفين منفصلين، مع الإشارة إلى البيانات التي تنتمي إلى أي مجموعة تصنيفية، أي crabs_testing/bold_Elasmobranchii.fasta و crabs_testing/bold_Mammalia.fasta .
for taxon in Elasmobranchii Mammalia; do crabs --download-bold --taxon ${taxon} --output crabs_testing/bold_${taxon}.fasta; done

خيار بديل: إلى جانب حلقة for الموصى بها، يمكن توفير أسماء أصناف متعددة مرة واحدة عن طريق فصل الأسماء باستخدام | .
crabs --download-bold --taxon 'Elasmobranchii|Mammalia' --output crabs_testing/bold_elasmobranchii_mammalia.fasta
--download-embl يتم تنزيل التسلسلات من EMBL من خلال موقع ENA FTP. سيتم أولاً تنزيل ملفات EMBL بتنسيق ".fasta.gz" وسيتم فك ضغطها تلقائيًا بمجرد اكتمال التنزيل. لا توفر قاعدة البيانات هذه قدرًا كبيرًا من المرونة فيما يتعلق بالتنزيل الانتقائي مقارنةً بـ BOLD أو NCBI. وبدلاً من ذلك، تم تنظيم بيانات EMBL في 15 قسمًا ضريبيًا، والتي يمكن تنزيلها بشكل منفصل. يمكن تحديد قسم الضريبة المراد تنزيله باستخدام المعلمة --taxon . نظرًا لأن كل قسم ضريبي مقسم إلى عدة ملفات، يتم توفير علامة * بعد الاسم لتنزيل كافة الملفات. يمكن للمستخدمين أيضًا تنزيل ملف معين عن طريق كتابة اسم الملف بالكامل. يتم توفير قائمة بجميع خيارات قسم الضرائب الخمسة عشر أدناه. يمكن تحديد دليل الإخراج واسم الملف باستخدام المعلمة --output .
قائمة الأقسام الضريبية:
crabs --download-embl --taxon 'mam*' --output crabs_testing/embl_mam.fasta
--download-mitofish يمكن لـ CRABS أيضًا تنزيل قاعدة بيانات MitoFish. قاعدة البيانات هذه عبارة عن ملف fasta واحد مكون من سطرين. يمكن تحديد دليل الإخراج واسم الملف باستخدام المعلمة --output .
crabs --download-mitofish --output crabs_testing/mitofish.fasta

--download-ncbi يتم تنزيل التسلسلات من قاعدة بيانات NCBI من خلال أدوات برمجة Entrez. يسمح NCBI بتنزيل البيانات من قواعد بيانات مختلفة، والتي يمكن للمستخدمين تحديدها باستخدام المعلمة --database . بالنسبة لمعظم المستخدمين، ستكون قاعدة بيانات --database nucleotide هي الأكثر ملاءمة لبناء قاعدة بيانات مرجعية محلية.
لتحديد البيانات التي سيتم تنزيلها من NCBI، يقوم المستخدمون بتوفير بحث من خلال المعلمة --query . قد يكون من الصعب صياغة عمليات بحث جيدة لـ NCBI. إحدى الطرق الجيدة لإنشاء استعلام بحث هي استخدام نافذة بحث صفحة ويب NCBI. من هذا الرابط، قم أولاً بإجراء بحث أولي ثم اضغط على زر الإدخال. سينقلك هذا إلى صفحة النتائج حيث يمكنك تحسين بحثك بشكل أكبر. في لقطة الشاشة أدناه، قمنا بتحسين البحث عن طريق الحد من طول التسلسل بين 100 - 25000 نقطة أساس ودمج تسلسلات الميتوكوندريا فقط. يمكن للمستخدمين نسخ النص ولصقه في مربع "تفاصيل البحث" على موقع الويب وتقديمه بين علامتي اقتباس إلى المعلمة --query . فائدة أخرى لاستخدام نافذة بحث صفحة الويب NCBI هي أن صفحة الويب ستعرض عدد التسلسلات التي تطابق استعلام البحث الخاص بك، والذي يجب أن يتطابق مع عدد التسلسلات التي أبلغ عنها CRABS. توفر صفحة الويب هذه برنامجًا تعليميًا قصيرًا إضافيًا حول استخدام وظيفة البحث على صفحة ويب NCBI التي كتبها فريقنا للحصول على معلومات إضافية.
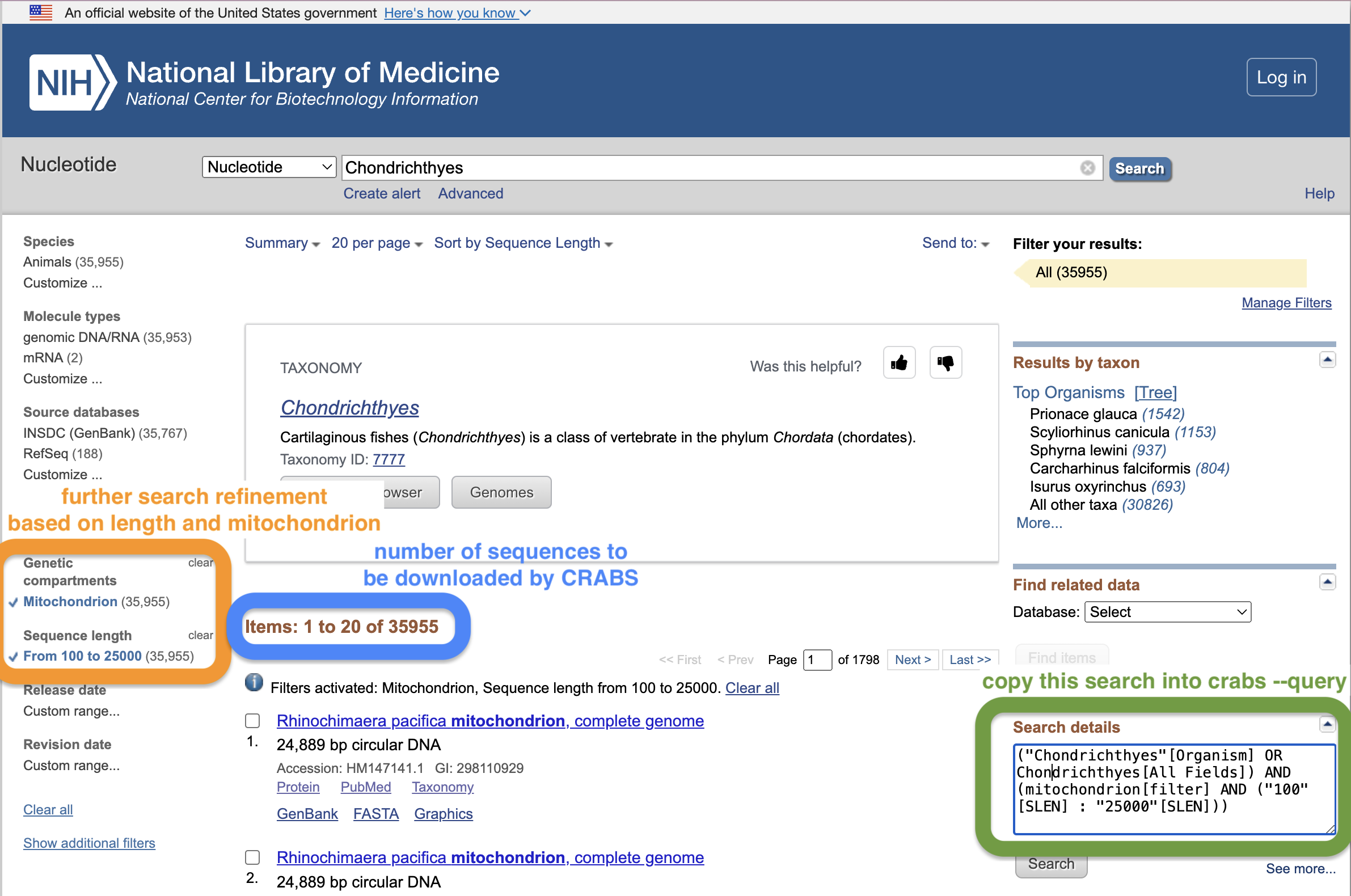
إلى جانب استعلام البحث ( --query )، يمكن للمستخدمين تقييد مصطلح البحث بشكل أكبر عن طريق تنزيل بيانات التسلسل لقائمة الأنواع باستخدام المعلمة --species . تأخذ المعلمة --species إما سلسلة إدخال لأسماء الأنواع مفصولة بعلامة + أو ملف إدخال بتنسيق txt يحتوي على اسم نوع واحد لكل سطر في المستند. توفر المعلمة --batchsize للمستخدمين خيار تنزيل التسلسلات على دفعات من N من موقع NCBI. هذه المعلمة الافتراضية هي 5000. لا يوصى بزيادة هذه القيمة فوق 5000، حيث من المرجح أن تقوم خوادم NCBI بفصل التنزيل إذا تم تنزيل عدد كبير جدًا من التسلسلات مرة واحدة. تتيح المعلمة --email للمستخدمين تحديد عنوان بريدهم الإلكتروني المطلوب للوصول إلى خوادم NCBI. وأخيرًا، يمكن تحديد دليل الإخراج واسم الملف باستخدام المعلمة --output .
crabs --download-ncbi --query '("Chondrichthyes"[Organism] OR Chondrichthyes[All Fields]) AND (mitochondrion[filter] AND ("100"[SLEN] : "25000"[SLEN]))' --output crabs_testing/ncbi_chondrichthyes.fasta --email [email protected] --database nucleotide

--import بمجرد تنزيل البيانات من المستودعات عبر الإنترنت، يجب استيراد الملفات إلى CRABS باستخدام وظيفة --import . يشكل تنسيق CRABS سطرًا واحدًا محددًا بعلامات جدولة لكل تسلسل يحتوي على جميع المعلومات، بما في ذلك (1) معرف التسلسل، (2) الاسم التصنيفي الذي تم تحليله من التنزيل الأولي، (3) رقم معرف تصنيف NCBI، (4) النسب التصنيفي وفقًا لتصنيف NCBI و (ت) التسلسل. سيحاول CRABS الحصول على رقم وصول NCBI لكل تسلسل كمعرف تسلسل. إذا كان التسلسل لا يحتوي على رقم وصول، أي لم يتم إيداعه في NCBI، فسيقوم CRABS بإنشاء معرفات تسلسل فريدة باستخدام التنسيق التالي: crabs_*[num]*_taxonomic_name . يتم تحديد تنسيق مستند الإدخال باستخدام المعلمة --import-format ويحدد اسم المستودع الذي تم تنزيل البيانات منه، على سبيل المثال، BOLD أو EMBL أو MITOFISH أو NCBI . يعتمد النسب التصنيفي الذي ينشئه CRABS على تصنيف NCBI ويتطلب CRABS الملفات الثلاثة التي تم تنزيلها باستخدام وظيفة --download-taxonomy ، أي --names و --nodes و --acc2tax . بدءًا من الإصدار v 1.0.0 ، أصبح CRABS قادرًا على حل الأسماء المترادفة والأسماء غير المقبولة لدمج عدد أكبر من التسلسلات والتنوع في قاعدة البيانات المرجعية المحلية. يمكن تحديد الرتب التصنيفية التي سيتم تضمينها في السلالة التصنيفية باستخدام معلمات --ranks . في حين أنه يمكن تضمين أي رتبة تصنيفية، فإننا نوصي باستخدام المدخلات التالية لتضمين جميع المعلومات الضرورية لمعظم المصنفات التصنيفية --ranks 'superkingdom;phylum;class;order;family;genus;species' . يمكن تحديد ملف الإخراج باستخدام المعلمة --output وهو ملف .txt بسيط. في النافذة الطرفية، يطبع CRABS نتائج عدد التسلسلات المستوردة، بالإضافة إلى أي تسلسلات لا يمكن إنشاء نسب تصنيفية لها.
crabs --import --import-format bold --input crabs_testing/bold_Elasmobranchii.fasta --names crabs_testing/names.dmp --nodes crabs_testing/nodes.dmp --acc2tax crabs_testing/nucl_gb.accession2taxid --output crabs_testing/crabs_bold.txt --ranks 'superkingdom;phylum;class;order;family;genus;species'

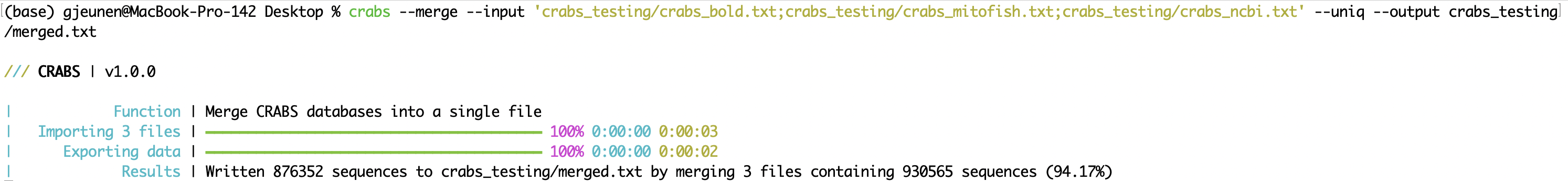
--merge عند تنزيل بيانات التسلسل من مستودعات متعددة عبر الإنترنت، يمكن دمج الملفات في ملف واحد بعد الاستيراد (راجع 5.2.1 --import ) باستخدام وظيفة --merge . يمكن إدخال ملفات الإدخال المراد دمجها باستخدام المعلمة --input ، مع فصل الملفات بـ ; . من الممكن أن يكون قد تم تنزيل التسلسل عدة مرات عند إيداعه في مستودعات مختلفة عبر الإنترنت. يؤدي استخدام المعلمة --uniq إلى الاحتفاظ بنسخة واحدة فقط من كل رقم وصول. يمكن تحديد ملف الإخراج باستخدام المعلمة --output . في نافذة المحطة الطرفية، يقوم CRABS بطباعة نتائج عدد التسلسلات المدمجة، بالإضافة إلى عدد التسلسلات التي تم الاحتفاظ بها عند استخدام المعلمة --uniq .
crabs --merge --input 'crabs_testing/crabs_bold.txt;crabs_testing/crabs_mitofish.txt;crabs_testing/crabs_ncbi.txt' --uniq --output crabs_testing/merged.txt
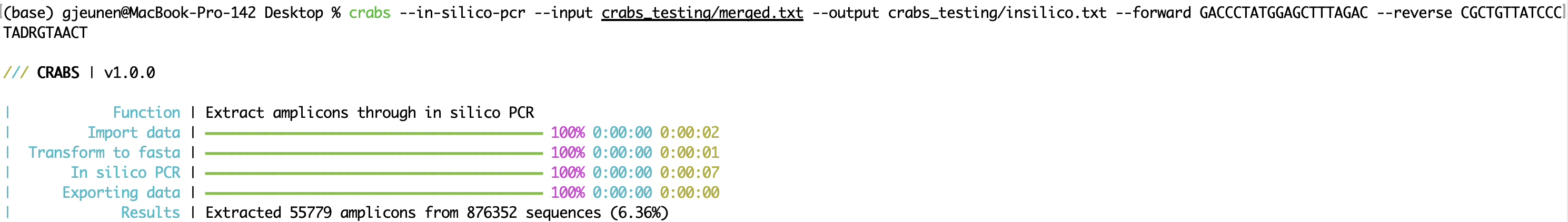
يستخرج CRABS منطقة amplicon من مجموعة التمهيدي عن طريق إجراء PCR في silico (الوظيفة: --in-silico-pcr ). يستخدم CRABS برنامج Cutadapt v 4.4 لـ in silico PCR لزيادة سرعة تنفيذ كود بايثون التقليدي. يمكن تحديد أسماء ملفات الإدخال والإخراج باستخدام المعلمات ' --input ' و' --output '، على التوالي. ينبغي توفير كل من التمهيدي الأمامي والخلفي في اتجاه 5'-3' باستخدام المعلمات ' --forward ' و' --reverse '، على التوالي. سوف يقوم CRABS بعكس مكمل التمهيدي العكسي. بدءًا من الإصدار v 1.0.0 ، أصبح CRABS قادرًا على الاحتفاظ بالرموز الشريطية في كلا الاتجاهين باستخدام تحليل واحد في silico PCR. ومن ثم، لا يتم إجراء أي خطوة تكامل عكسية وإعادة تشغيل PCR في silico ، وبالتالي زيادة سرعة التنفيذ بشكل ملحوظ. للاحتفاظ بالتسلسلات التي لا يمكن العثور على مناطق ربط أولية لها، يمكن تحديد ملف إخراج للمعلمة --untrimmed . يمكن تحديد الحد الأقصى المسموح به لعدد حالات عدم التطابق الموجودة في مناطق الربط التمهيدي باستخدام المعلمة --mismatch ، مع الإعداد الافتراضي 4. وأخيرًا، يمكن أن يكون تحليل PCR في silico متعدد الخيوط في CRABS. افتراضيًا، يتم استخدام الحد الأقصى لعدد سلاسل الرسائل، ولكن يمكن للمستخدمين تحديد عدد سلاسل الرسائل المراد استخدامها مع المعلمة --threads .
crabs --in-silico-pcr --input crabs_testing/merged.txt --output crabs_testing/insilico.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT
من الممارسات الشائعة إزالة مناطق الربط التمهيدي من التسلسلات المرجعية عند إيداعها في قاعدة بيانات على الإنترنت. لذلك، عندما يتم إنشاء التسلسل المرجعي باستخدام نفس التمهيدي الأمامي و/أو العكسي كما تم البحث عنه في الدالة --in-silico-pcr ، ستفشل الدالة --in-silico-pcr في استرداد منطقة amplicon الخاصة بالـ التسلسل المرجعي. لمراعاة هذا الاحتمال، لدى CRABS خيار تشغيل محاذاة عالمية زوجية، يتم تنفيذها باستخدام VSEARCH v 2.16.0 ، لاستخراج مناطق amplicon التي لا يحتوي التسلسل المرجعي لها على مناطق الربط التمهيدي الأمامية والعكسية الكاملة. لتحقيق ذلك، تأخذ الدالة --pairwise-global-alignment ملف قاعدة البيانات الذي تم تنزيله في الأصل باستخدام المعلمة --input . قاعدة البيانات التي سيتم البحث عنها هي ملف الإخراج من --in-silico-pcr ويمكن تحديدها باستخدام المعلمة --amplicons . يمكن تحديد ملف الإخراج باستخدام المعلمة --output . يمكن ضبط التسلسلات التمهيدية، المستخدمة فقط لحساب طول الزوج الأساسي، باستخدام المعلمات --forward و --reverse . نظرًا لأن وظيفة --pairwise-global-alignment يمكن أن تستغرق وقتًا طويلاً للتشغيل في قواعد البيانات الكبيرة، فيمكن تقييد طول التسلسل لتسريع العملية باستخدام المعلمة --size-select . يمكن تحديد الحد الأدنى لنسبة الهوية وتغطية الاستعلام باستخدام معلمات --percent-identity و --coverage ، على التوالي. يجب تقديم --percent-identity كقيمة مئوية بين 0 و1 (على سبيل المثال، 95% = 0.95)، بينما يجب تقديم --coverage كقيمة مئوية بين 0 و100 (على سبيل المثال، 95% = 95). افتراضيًا، تقتصر وظيفة --pairwise-global-alignment على الاحتفاظ بالتسلسلات التي لا تكون فيها التسلسلات التمهيدية موجودة بشكل كامل في التسلسل المرجعي (تبدأ المحاذاة أو تنتهي ضمن طول التمهيدي الأمامي أو العكسي). عند توفير المعلمة --all-start-positions ، سيتم تضمين النتائج الإيجابية عندما يتم العثور على المحاذاة خارج نطاق مناطق الربط التمهيدي (التي تفتقدها الدالة --in-silico-pcr بسبب وجود عدد كبير جدًا من حالات عدم التطابق في منطقة الربط التمهيدي). لا نوصي باستخدام --all-start-positions ، لأنه من غير المرجح أن يتم تضخيم الرمز الشريطي باستخدام مجموعة التمهيدي المحددة للدالة --in-silico-pcr عند وجود أكثر من 4 حالات عدم تطابق في التمهيدي- مناطق ملزمة.
crabs --pairwise-global-alignment --input crabs_testing/merged.txt --amplicons crabs_testing/insilico.txt --output crabs_testing/aligned.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT --size-select 10000 --percent-identity 0.95 --coverage 0.95
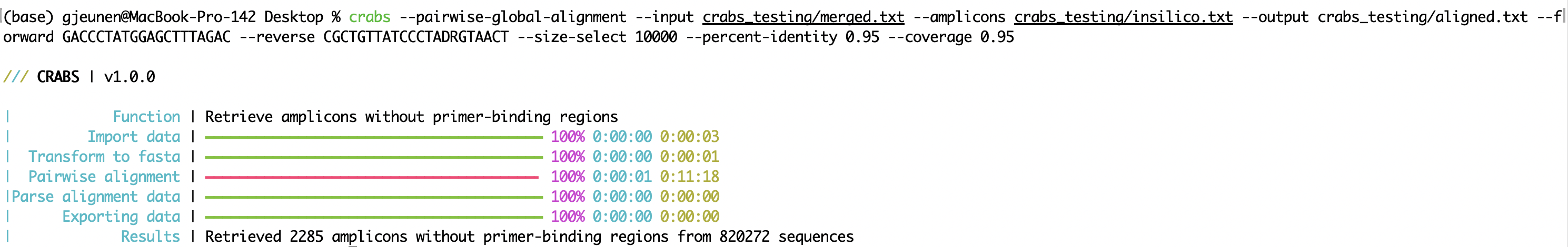
--pairwise-global-alignment يمكن أن تستغرق وظيفة --pairwise-global-alignment قدرًا كبيرًا من الوقت للتنفيذ عندما يقوم CRABS بمعالجة ملفات تسلسلية كبيرة، على الرغم من دعم مؤشرات الترابط المتعددة. منذ التحديث إلى CRABS v 1.0.0 ، تم وضع بنية ملف متطابقة من --import إلى --export ، وبالتالي تمكين تنفيذ الوظائف بأي ترتيب. على الرغم من أننا لا نزال نوصي باتباع ترتيب سير عمل CRABS، إلا أنه يمكن تسريع وظيفة --pairwise-global-alignment بشكل ملحوظ عند تنفيذ وظائف --dereplicate و --filter قبل وظيفة --in-silico-pcr . من خلال تنفيذ خطوات التنظيم هذه قبل --in-silico-pcr ، سيتم تقليل عدد التسلسلات اللازمة للمعالجة بواسطة CRABS لوظيفة --pairwise-global-alignment بشكل كبير.
الملاحظة 1 : عند تنفيذ وظيفة --filter قبل --in-silico-pcr ، يرجى التأكد من حذف أي معلمات تؤثر بشكل مباشر على التسلسل، حيث أن --filter سيبني هذا على التسلسل بأكمله وليس على amplicon المستخرج . ومن ثم، قم بحذف المعلمات التالية: --minimum-length , --maximum-length , --maximum-n .
الملاحظة 2 : عند تنفيذ وظائف --dereplicate و --filter قبل --in-silico-pcr ، سيكون من المستحسن تشغيل كلتا الوظيفتين مرة أخرى بعد --pairwise-global-alignment ، حيث يمكن تنظيم قاعدة البيانات بشكل أكبر الآن أن يتم استخراج amplicons.
بمجرد استخراج جميع الرموز الشريطية المحتملة لمجموعة التمهيدي بواسطة وظائف --in-silico-pcr و-- --pairwise-global-alignment ، يمكن لقاعدة البيانات المرجعية المحلية أن تخضع لمزيد من التنظيم والإعداد الفرعي داخل CRABS باستخدام وظائف مختلفة، بما في ذلك --dereplicate و --filter و --subset .
--dereplicate الطريقة الأولى للتنظيم هي إلغاء تكرار قاعدة البيانات المرجعية المحلية باستخدام الدالة --dereplicate . من الممكن أنه بالنسبة لبعض الأصناف، يتم تضمين العديد من الرموز الشريطية المتطابقة في قاعدة البيانات المرجعية المحلية في هذه المرحلة. يمكن أن يحدث هذا عندما تقوم مجموعات بحثية مختلفة بإيداع تسلسلات متطابقة أو إذا لم يكن التباين الداخلي بين تسلسلات أحد الأصنوفة موجودًا في الباركود المستخرج. من الأفضل إزالة هذه الرموز الشريطية المرجعية المتطابقة لتسريع مهمة التصنيف، بالإضافة إلى تحسين نتائج مهمة التصنيف (خاصة بالنسبة للمصنفات التصنيفية التي توفر عددًا محدودًا من النتائج، على سبيل المثال، BLAST).
يمكن تحديد ملفات الإدخال والإخراج باستخدام المعلمات --input و --output ، على التوالي. يقدم CRABS ثلاث طرق لإلغاء النسخ المتماثل، والتي يمكن تحديدها باستخدام معلمة --dereplication-method ، بما في ذلك:
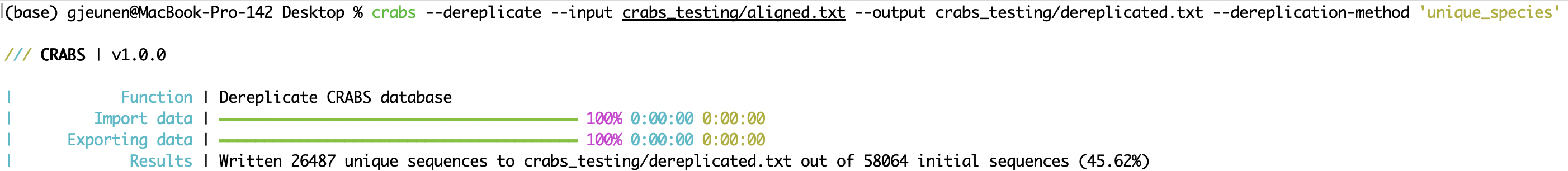
crabs --dereplicate --input crabs_testing/aligned.txt --output crabs_testing/dereplicated.txt --dereplication-method 'unique_species'
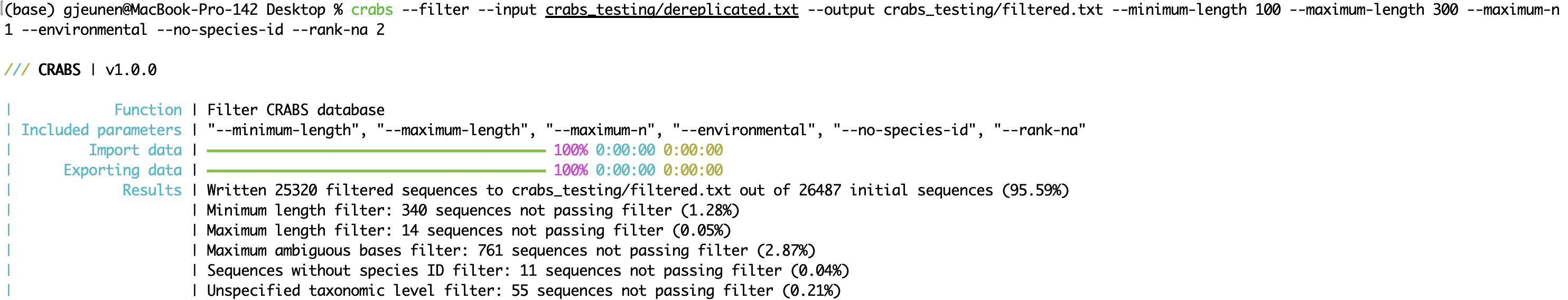
--filter طريقة التنظيم الثانية هي تصفية قاعدة البيانات المرجعية المحلية باستخدام معلمات مختلفة باستخدام وظيفة --filter . يمكن تحديد ملفات الإدخال والإخراج باستخدام المعلمات --input و --output ، على التوالي. من الإصدار الخامس 1.0.0 . يتضمن CRABS التصفية بناءً على ستة معلمات، بما في ذلك:
--minimum-length : الحد الأدنى لطول التسلسل الذي سيتم الاحتفاظ به في قاعدة البيانات.--maximum-length : الحد الأقصى لطول التسلسل الذي سيتم الاحتفاظ به في قاعدة البيانات.--maximum-n : تجاهل amplicons مع N أو أكثر من القواعد الغامضة ( N ) ؛--environmental : تجاهل التسلسل البيئي من قاعدة البيانات؛--no-species-id : تجاهل التسلسلات التي لا يتوفر لها اسم نوع؛--rank-na : تجاهل التسلسلات التي تحتوي على N أو أكثر من المستويات التصنيفية غير المحددة. crabs --filter --input crabs_testing/dereplicated.txt --output crabs_testing/filtered.txt --minimum-length 100 --maximum-length 300 --maximum-n 1 --environmental --no-species-id --rank-na 2
--subset تتمثل طريقة التنظيم الثالثة والأخيرة المضمنة في CRABS في تعيين قاعدة بيانات مرجعية محلية فرعية لتضمين (المعلمة: --include ) أو استبعاد (المعلمة: --exclude ) تصنيفات محددة باستخدام وظيفة --subset . تسمح هذه الوظيفة بإزالة الرموز الشريطية المرجعية من المجموعات التصنيفية التي لا تهم سؤال البحث. كان من الممكن دمج هذه المجموعات التصنيفية في قاعدة البيانات المرجعية المحلية بسبب التضخيم المحتمل غير المحدد للمجموعة التمهيدية. حالة استخدام أخرى لـ --subset هي إزالة التسلسلات الخاطئة المعروفة.
بالنسبة للمصنفات التصنيفية المستندة إلى التعلم الآلي (IDTAXA) أو مسافة k-mer (SINTAX)، قد يكون من المفيد تقسيم قاعدة البيانات المرجعية عن طريق تضمين الأصناف المعروفة بحدوثها فقط في المنطقة التي تم فيها أخذ العينات واستبعاد الأنواع ذات الصلة الوثيقة المعروفة التي ستحدث في المنطقة لزيادة الدقة التصنيفية التي تم الحصول عليها لهذه المصنفات والحصول على نتائج محسنة لتخصيص التصنيف.
يمكن تحديد ملفات الإدخال والإخراج باستخدام المعلمات --input و --output ، على التوالي. يمكن أن تحتوي المعلمات --include و --exclude على قائمة من الأصناف مفصولة بـ ; أو ملف .txt يحتوي على اسم تصنيف واحد في كل سطر.
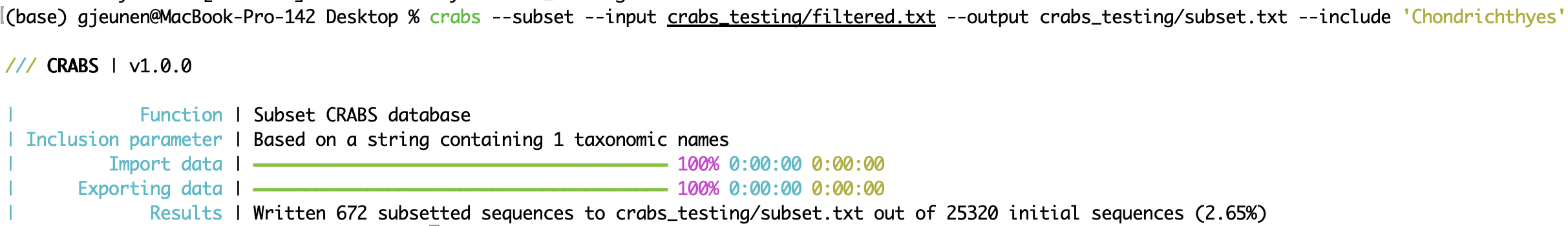
crabs --subset --input crabs_testing/filtered.txt --output crabs_testing/subset.txt --include 'Chondrichthyes'
بمجرد الانتهاء من قاعدة البيانات المرجعية، يمكن تصديرها إلى تنسيقات مختلفة لاستيعاب المواصفات التي تتطلبها معظم الأدوات البرمجية التي تخصص التصنيف للبيانات الميتاجينومية. يمكن تحديد ملفات الإدخال والإخراج باستخدام المعلمات --input و --output ، على التوالي. من الإصدار v 1.0.0 ، قام CRABS بدمج تنسيق قاعدة البيانات المرجعية لستة مصنفات مختلفة (المعلمة: --export-format )، بما في ذلك:
--export-format 'sintax' : تم دمج مصنف SINTAX في USEARCH وVSEARCH؛--export-format 'rdp' : مُصنف RDP هو برنامج مستقل يستخدم على نطاق واسع في دراسات الميكروبيوم؛--export-format 'qiime-fasta' و- --export-format 'qiime-text' : يمكن استخدامها لتعيين معرف تصنيفي في QIIME وQIIME2؛--export-format 'dada2-species' و- --export-format 'dada2-taxonomy' : يمكن استخدامها لتعيين معرف تصنيفي في DADA2؛--export-format 'idt-fasta' و- --export-format 'idt-text' : مصنف IDTAXA عبارة عن خوارزمية تعلم آلي مدمجة في حزمة DECIPHER R؛--export-format 'blast-notax' : ينشئ قاعدة بيانات مرجعية محلية للانفجار لـ BLASTN و MEGABLAST حيث لا يوفر الإخراج معرفًا تصنيفيًا ، ولكنه يسرد رقم الانضمام ؛--export-format 'blast-tax' : إنشاء قاعدة بيانات مرجعية محلية للانفجار لـ BLASTN و MEGABLAST حيث يوفر الإخراج كل من معرف التصنيف ورقم الانضمام. crabs --export --input crabs_testing/subset.txt --output crabs_testing/BLAST_TAX_CHONDRICHTHYES --export-format 'blast-tax'

أثناء تصدير قاعدة البيانات المرجعية المحلية إلى تنسيق واحد (باستثناء المصنفات التي يتم فيها تقسيم قاعدة البيانات المرجعية عبر ملفات متعددة ، فإن Qiime و DADA2 و IDTAXA) تكفي لمعظم المستخدمين ، ويمكن كتابة بسيطة للحلقة لتصدير المحلية قاعدة البيانات المرجعية لتنسيقات متعددة إذا كان المستخدمون يرغبون في مقارنة النتائج بين المصنفات التصنيفية المختلفة. تم توفير مثال أدناه لتصدير قاعدة بيانات المرجع المحلية بتنسيقات Sintax و RDP و Idtaxa.
for format in sintax.fasta rdp.fasta idt-fasta.fasta idt-text.txt; do crabs --export --input crabs_testing/subset.txt --output crabs_testing/chondrichthyes_${format} --export-format ${format%%.*}; done

بمجرد الانتهاء من قاعدة البيانات المرجعية ، يمكن لسرطان البحر تشغيل خمس وظائف بعد المعالجة لاستكشاف وتوفير نظرة عامة موجزة على قاعدة البيانات المرجعية المحلية ، بما في ذلك (I) --diversity-figure ، (II) --amplicon-length-figure ، (( (3) --phylogenetic-tree ، (4) --amplification-efficiency-figure ، و (V) --completeness-table .
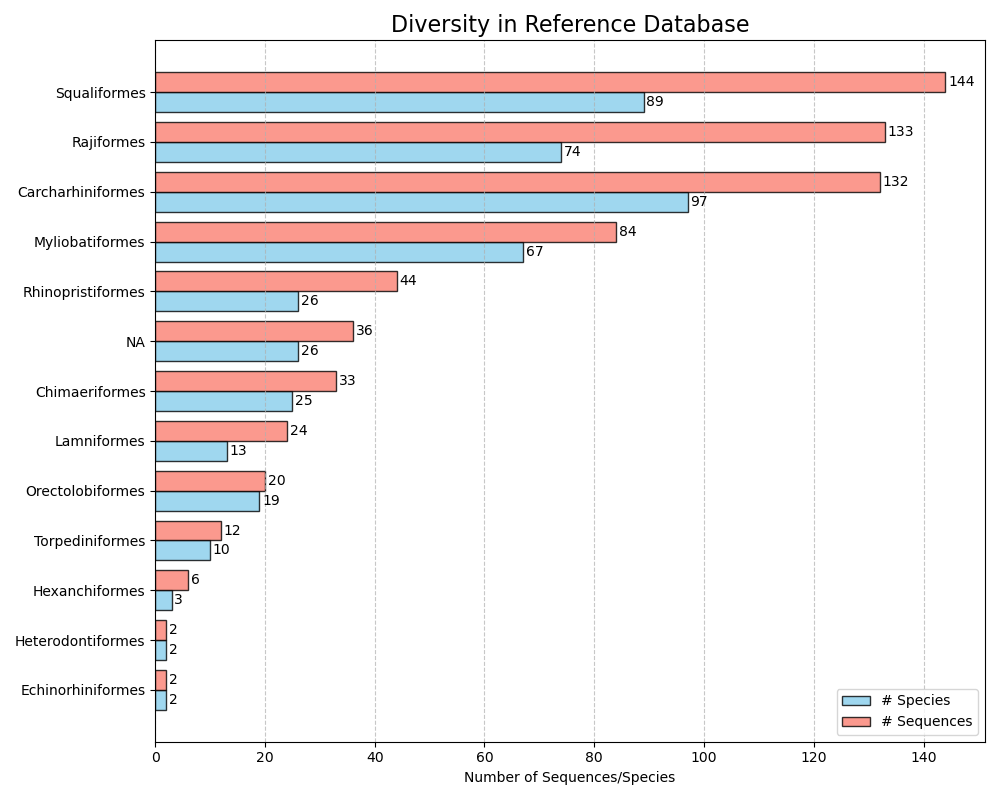
--diversity-figure تنتج --diversity-figure ، مؤامرة شريط أفقي مع عدد من الأنواع (باللون الأزرق) وعدد التسلسل (باللون البرتقالي) لكل مجموعة تصنيفية في قاعدة البيانات المرجعية. يمكن للمستخدم تحديد المرتبة التصنيفية لتقسيم قاعدة بيانات المرجع بالمعلمة على --tax-level . المستوى الضريبي هو عدد المرتبة التي ظهر فيها خلال وظيفة --import . على سبيل المثال ، إذا --ranks 'superkingdom;phylum;class;order;family;genus;species' خلال --import فسيجد الانقسام استنادًا إلى superkingdom-على --tax-level 1 ، phylum = --tax-level 2 ، class = --tax-level 3 ، إلخ. يمكن تحديد ملف الإدخال بتنسيق سرطان البحر باستخدام المعلمة --input . سيتم كتابة الشكل ، بتنسيق .png ، إلى ملف الإخراج ، والذي يمكن تحديده باستخدام المعلمة --output .
crabs --diversity-figure --input crabs_testing/subset.txt --output crabs_testing/diversity-figure.png --tax-level 4
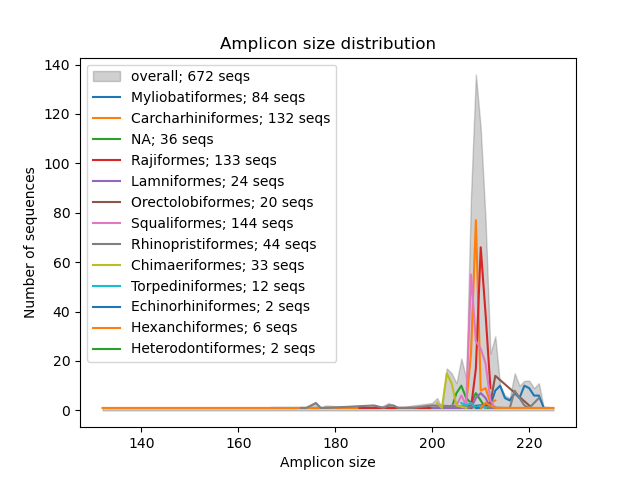
--amplicon-length-figure تنتج وظيفة --amplicon-length-figure رسمًا بيانيًا خطًا يعرض نطاق طول Amplicon. يتم عرض النطاق الإجمالي في طول Amplicon عبر جميع التسلسلات في قاعدة البيانات المرجعية باللون الرمادي المظلل ، في حين أن النتائج تنقسم لكل مجموعة تصنيفية (المعلمة: --tax-level ) متداخلة بواسطة خطوط ملونة. بالإضافة إلى ذلك ، يعرض الأسطورة عدد التسلسلات المعينة لكل من المجموعات التصنيفية وإجمالي عدد التسلسلات في قاعدة البيانات المرجعية. يمكن تحديد ملف الإدخال بتنسيق سرطان البحر باستخدام المعلمة --input . سيتم كتابة الشكل ، بتنسيق .png ، إلى ملف الإخراج ، والذي يمكن تحديده باستخدام المعلمة --output .
crabs --amplicon-length-figure --input crabs_testing/subset.txt --output crabs_testing/amplicon-length-figure.png --tax-level 4
--phylogenetic-tree ستولد وظيفة --phylogenetic-tree الشجرة شجرة phylogenetic لقائمة من الأنواع ذات الاهتمام. يمكن استيراد قائمة الأنواع ذات الاهتمام باستخدام المعلمة --species وتتكون إما من سلسلة إدخال مفصولة بواسطة ملف + txt مع اسم نوع واحد على كل سطر. لكل نوع من الفائدة ، سيتم استخراج التسلسلات من قاعدة البيانات المرجعية التي تشترك في رتبة تصنيفية محددة من قبل المستخدم (المعلمة: --tax-level ) مع الأنواع ذات الاهتمام. سوف يولد السلطعون محاذاة جميع التسلسلات المستخرجة باستخدام clustalw2 v 2.1 وتوليد شجرة phylogenetic التي تنضج الجوار باستخدام Fasttree. ستتم كتابة هذه الشجرة التطورية بتنسيق Newick إلى ملف الإخراج باستخدام المعلمة --output ويمكن تصورها في برامج البرامج مثل Figtree أو الجينات. نظرًا لأن شجرة phylogenetic منفصلة سيتم إنشاء كل نوع من المهمة ، --output يأخذ اسم ملف عام ، في حين أن ملف الإخراج الدقيق سيحتوي على هذا الاسم العام متبوعًا بـ '_species_name.tree'.
crabs --phylogenetic-tree --input crabs_testing/subset.txt --output crabs_testing/phylo --tax-level 4 --species 'Carcharodon carcharias+Squalus acanthias'
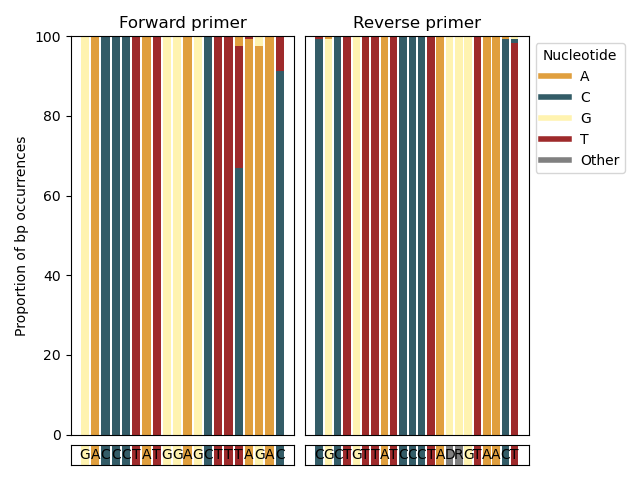
--amplification-efficiency-figure ستنتج وظيفة --amplification-efficiency-figure رسم بياني شريطية ، يعرض نسبة حدوث زوج أساسي في المناطق المرتبطة بالتمهيدي لمجموعة تصنيفية محددة للمستخدم ، وبالتالي تصور الأماكن في المناطق الأمامية والخلفية للربط التمهيدي حيث لا تتطابق مع عدم التطابق قد يحدث في مجموعة الفائدة التصنيفية ، مما قد يؤثر على كفاءة التضخيم. تأخذ وظيفة --amplification-efficiency-figure قاعدة بيانات مرجعية تنسيقها CARBS النهائية كمدخلات باستخدام المعلمة- --amplicons . للعثور على المعلومات الخاصة بالمناطق المرتبطة بالتمهيدي لكل تسلسل في ملف الإدخال ، يجب توفير التسلسلات التي تم تنزيلها في البداية بعد الاستيراد باستخدام المعلمة --input . يتم توفير تسلسل التمهيدي للأمام والعكس (في اتجاه 5 ' -3') باستخدام المعلمات --forward و --reverse . يمكن توفير اسم مجموعة الفائدة التصنيفية باستخدام معلمة --tax-group ويمكن تعيينها على أي مستوى تصنيفي تم دمجه في ملف الإدخال. أخيرًا ، سيتم كتابة الشكل بتنسيق .png إلى ملف الإخراج المحدد بواسطة المعلمة --output .
crabs --amplification-efficiency-figure --input crabs_testing/merged.txt --amplicons crabs_testing/subset.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT --output crabs_testing/amplification-efficiency.png --tax-group Carcharhiniformes
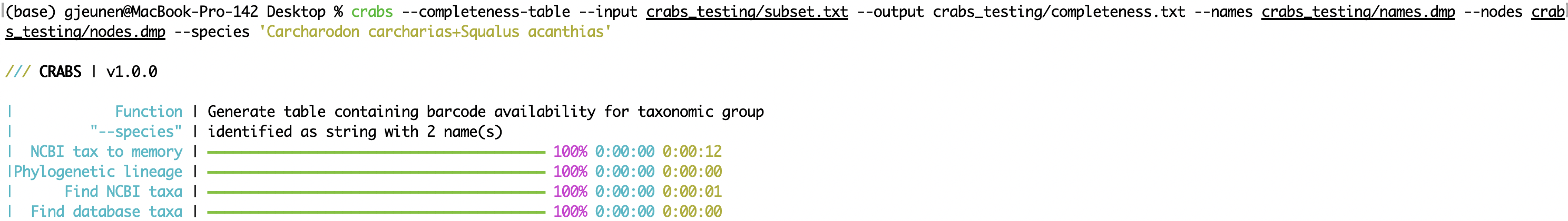
--completeness-table ستؤدي وظيفة --completeness-table إلى إخراج جدول علامات التبويب (المعلمة: --output ) مع معلومات حول قائمة الأنواع ذات الاهتمام. يمكن استيراد قائمة الأنواع ذات الاهتمام باستخدام المعلمة --species وتتكون إما من سلسلة إدخال مفصولة بواسطة ملف + txt مع اسم نوع واحد على كل سطر. --names إنشاء نسب تصنيفية لكل نوع --download-taxonomy الاهتمام باستخدام --nodes " الأسماء . سيكون لجدول الإخراج 10 أعمدة توفر المعلومات التالية:
crabs --completeness-table --input crabs_testing/subset.txt --output crabs_testing/completeness.txt --names crabs_testing/names.dmp --nodes crabs_testing/nodes.dmp --species 'Carcharodon carcharias+Squalus acanthias'
crabs --version v 1.0.6 : إصلاح الأخطاء -> تحسين تحليل الرؤوس الجريئة أثناء --import .crabs --version v 1.0.5 : Press Fix -> أضاف قيود الطول إلى معرف SEQ عند إنشاء قواعد بيانات الانفجار ، حسب الحاجة لبرنامج Blast+.crabs --version v 1.0.4 : Inday Info-> قدمت المعلومات الصحيحة حول إدخال القيمة لـ- --pairwise-global-alignment --coverage --percent-identity .crabs --version v 1.0.3 : إصلاح الأخطاء -> التحقق من استجابة خادم NCBI 3 مرات قبل إحباط التحليل.crabs --version v 1.0.2 : إصلاح الأخطاء -> قادر على الإبلاغ عند إرجاع تسلسل 0 بعد التحليل.crabs --version v 1.0.1 : Bug Fix -> Building Building NCBI Query باستخدام المعلمة --species .

