nucleotide transformer
1.0.0
مرحبًا بك في مستودع InstaDeep Github، حيث يتم عرض ما يلي:
يسعدنا فتح هذه الأعمال وتزويد المجتمع بإمكانية الوصول إلى الكود والأوزان المدربة مسبقًا لنماذج لغة الجينوم التسعة ونموذجي التجزئة. تم تطوير نماذج من مشروع nucleotide transformer بالتعاون مع Nvidia وTUM، وتم تدريب النماذج على عقد DGX A100 على Cambridge-1. تم تطوير النموذج من مشروع nucleotide transformer الزراعية بالتعاون مع Google، وتم تدريب النموذج على مسرعات TPU-v4.
بشكل عام، توفر أعمالنا رؤى جديدة تتعلق بالتدريب المسبق وتطبيق النماذج التأسيسية للغة، بالإضافة إلى تدريب النماذج التي تستخدمها كمشفر أساسي لعلم الجينوم مع فرص كبيرة لتطبيقاتها في هذا المجال.
ستجد في هذا المستودع ما يلي:
بالمقارنة مع الأساليب الأخرى، فإن نماذجنا لا تدمج المعلومات من الجينومات المرجعية الفردية فحسب، بل تستفيد من تسلسل الحمض النووي من أكثر من 3200 جينوم بشري متنوع، بالإضافة إلى 850 جينومًا من مجموعة واسعة من الأنواع، بما في ذلك الكائنات الحية النموذجية وغير النموذجية. من خلال تقييم قوي وموسع، نظهر أن هذه النماذج الكبيرة توفر تنبؤًا دقيقًا للغاية بالنمط الظاهري الجزيئي مقارنة بالطرق الحالية.
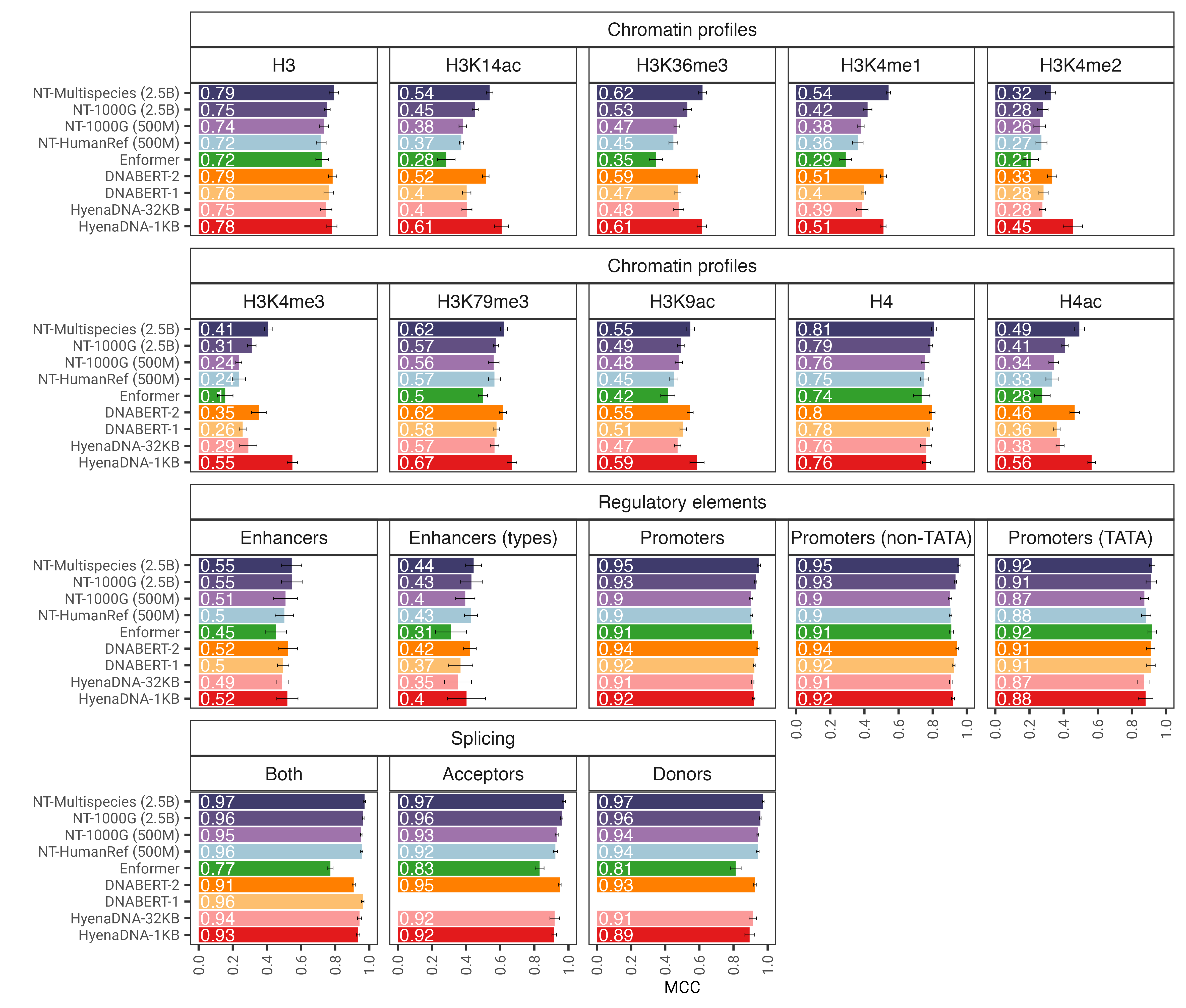
الشكل 1: يتنبأ نموذج nucleotide transformer بدقة بمهام الجينوم المتنوعة بعد الضبط الدقيق. نعرض نتائج الأداء عبر المهام النهائية لنماذج المحولات المضبوطة بدقة. تمثل أشرطة الخطأ 2 SDs مشتقة من التحقق المتبادل بمقدار 10 أضعاف.
نقدم في هذا العمل نموذجًا أساسيًا جديدًا للغة كبيرة تم تدريبه على الجينومات المرجعية من 48 نوعًا نباتيًا مع التركيز السائد على أنواع المحاصيل. قمنا بتقييم أداء AgroNT عبر العديد من مهام التنبؤ التي تتراوح بين الميزات التنظيمية، ومعالجة الحمض النووي الريبي (RNA)، والتعبير الجيني، وأظهرنا أن AgroNT يمكنه الحصول على أداء متطور.
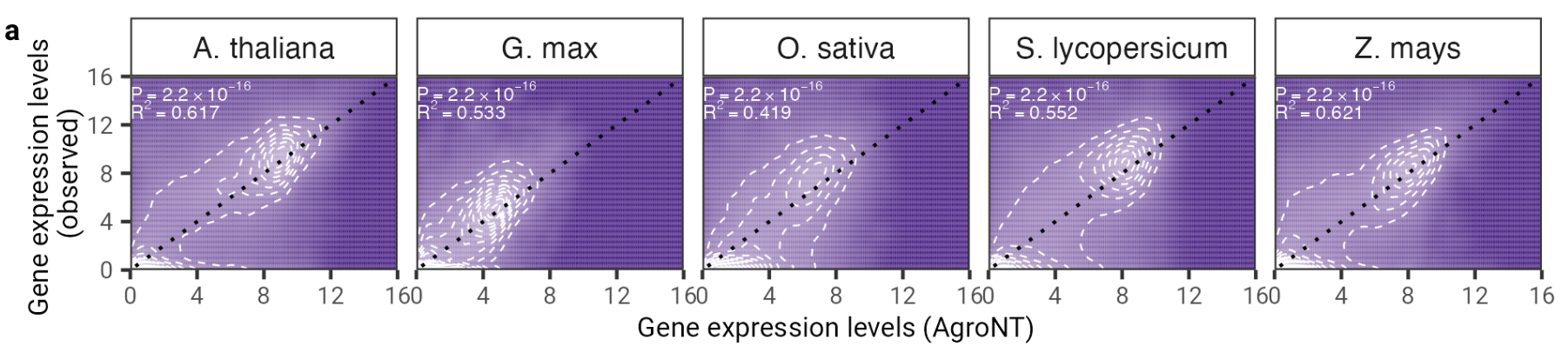
الشكل 2: يوفر AgroNT تنبؤًا بالتعبير الجيني عبر أنواع نباتية مختلفة. يرتبط التنبؤ بالتعبير الجيني على الجينات المقاومة في جميع الأنسجة بمستويات التعبير الجيني المرصودة. يتم عرض معامل التحديد (R 2 ) من النموذج الخطي والقيم P المرتبطة به بين القيم المتوقعة والمرصودة.
لاستخدام الكود والنماذج المدربة مسبقًا، ما عليك سوى:
pip install . .يمكنك بعد ذلك تنزيل أي من نماذجنا التسعة وإجراء الاستدلال عليها في بضعة أسطر فقط من الرموز:
import haiku as hk
import jax
import jax . numpy as jnp
from nucleotide_transformer . pretrained import get_pretrained_model
# Get pretrained model
parameters , forward_fn , tokenizer , config = get_pretrained_model (
model_name = "500M_human_ref" ,
embeddings_layers_to_save = ( 20 ,),
max_positions = 32 ,
)
forward_fn = hk . transform ( forward_fn )
# Get data and tokenize it
sequences = [ "ATTCCGATTCCGATTCCG" , "ATTTCTCTCTCTCTCTGAGATCGATCGATCGAT" ]
tokens_ids = [ b [ 1 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens_str = [ b [ 0 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens = jnp . asarray ( tokens_ids , dtype = jnp . int32 )
# Initialize random key
random_key = jax . random . PRNGKey ( 0 )
# Infer
outs = forward_fn . apply ( parameters , random_key , tokens )
# Get embeddings at layer 20
print ( outs [ "embeddings_20" ]. shape )أسماء النماذج المدعومة هي:
يمكنك أيضًا تشغيل نماذجنا والعثور على مزيد من أمثلة التعليمات البرمجية في google colab
يعمل الكود على GPU وTPU بفضل Jax!
تشتمل نماذج الإصدار الثاني nucleotide transformer الخاصة بنا على سلسلة من التغييرات المعمارية التي أثبتت أنها أكثر كفاءة: بدلاً من استخدام التضمينات الموضعية المستفادة، نستخدم التضمينات الدوارة التي يتم استخدامها في كل طبقة انتباه ووحدات خطية مسورة مع عمليات تنشيط حفيف دون تحيز. تقبل هذه النماذج المحسنة أيضًا تسلسلات تصل إلى 2048 رمزًا مميزًا مما يؤدي إلى نافذة سياق أطول تبلغ 12 كيلو بايت في الثانية. مستوحاة من قوانين تحجيم شينشيلا، قمنا أيضًا بتدريب نماذج NT-v2 الخاصة بنا على مجموعة البيانات متعددة الأنواع لدينا لمدة أطول (300 مليار رمز مميز لنماذج 50M و100M؛ رموز 1T لنموذج 250M و500M) مقارنة بنماذج v1 (300B رمز مميز لجميع النماذج الأربعة).
طبقات المحولات مفهرسة بـ 1، مما يعني أن استدعاء get_pretrained_model باستخدام الوسيطات model_name="500M_human_ref" و embeddings_layers_to_save=(1, 20,) سيؤدي إلى استخراج التضمينات بعد طبقة المحولات الأولى والعشرون. بالنسبة للمحولات التي تستخدم رأس Roberta LM، من الشائع استخراج التضمينات النهائية بعد معيار الطبقة الأولى لرأس LM بدلاً من بعد كتلة المحولات الأخيرة. لذلك، إذا تم استدعاء get_pretrained_model باستخدام الوسيطات التالية embeddings_layers_to_save=(24,) ، فلن يتم استخراج التضمينات بعد طبقة المحول النهائية بل بعد معيار الطبقة الأولى لرأس LM.
تستفيد نماذج SegmentNT من محول nucleotide transformer (NT) الذي أزلنا منه رأس نموذج اللغة واستبدلناه برأس تجزئة U-Net أحادي البعد للتنبؤ بموقع عدة أنواع من عناصر الجينوم في تسلسل بدقة نيوكليوتيد واحدة. نقدم نموذجين مختلفين على 14 فئة مختلفة من عناصر الجينوم البشري في تسلسلات إدخال تصل إلى 30 كيلو بايت. وتشمل هذه الجينات (جينات ترميز البروتين، وlncRNAs، و5'UTR، و3'UTR، وexon، وintron، ومتقبلات الوصلات، والمواقع المانحة) والتنظيمية (إشارة polyA، ومحفزات ومعززات الأنسجة الثابتة والمحددة للأنسجة، وCTCF المرتبطة المواقع) العناصر. يحقق SegmentNT أداءً فائقًا مقارنة بأحدث بنية تجزئة U-Net، مستفيدًا من أوزان NT المدربة مسبقًا، ويوضح تعميمًا بدون إطلاق يصل إلى 50 كيلو بايت في الثانية.
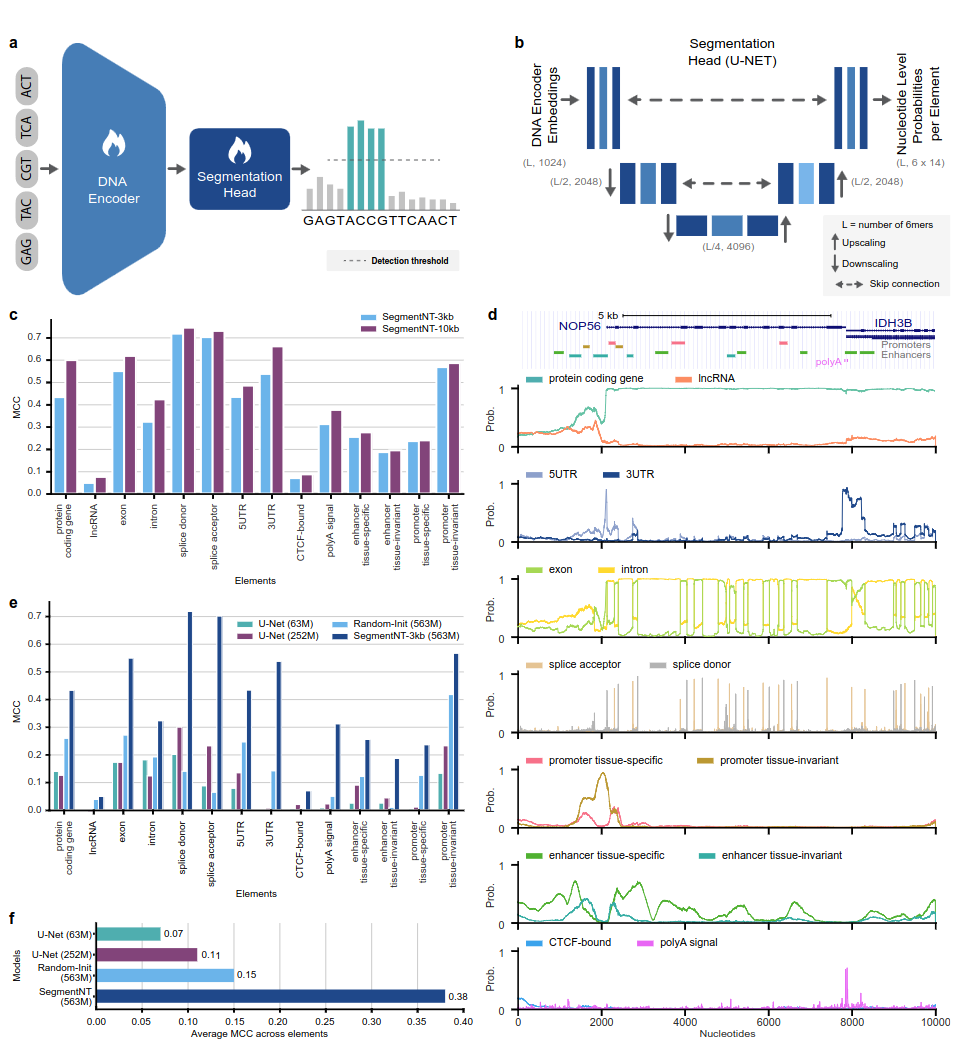
الشكل 1: يقوم SegmentNT بتوطين عناصر الجينوم بدقة النوكليوتيدات.
لاستخدام الكود والنماذج المدربة مسبقًا، ما عليك سوى:
pip install . .يمكنك بعد ذلك تنزيل التسلسل واستنتاجه باستخدام أي من نماذجنا في بضعة أسطر فقط من الرموز:
rescaling factor على العامل المستخدم أثناء التدريب. في حالة رغبتك في استنتاج تسلسلات بين 30 كيلو بايت و50 كيلو بايت، تأكد من تمرير وسيطة rescaling_factor في وظيفة get_pretrained_segment_nt_model بالقيمة rescaling_factor = max_num_nucleotides / max_num_tokens_nt حيث num_dna_tokens_inference هو عدد الرموز المميزة عند الاستدلال (أي 6669 لتسلسل من 40008 زوجًا أساسيًا) و max_num_tokens_nt هو الحد الأقصى لعدد الرموز التي تم تدريب محول النوكليوتيدات الأساسي عليها، أي 2048 .
؟ يعرض examples/inference_segment_nt.ipynb كيفية الاستدلال على تسلسل بحجم 50 كيلو بايت ورسم احتمالات إعادة إنتاج الشكل 3 من الورقة.
؟ لا تتعامل نماذج SegmentNT مع أي "N" في تسلسل الإدخال لأن كل نيوكليوتيدات تحتاج إلى ترميزها على أنها 6-mers، وهو ما لا يمكن أن يكون هو الحال عند استخدام تسلسلات تحتوي على زوج أساسي واحد أو عدة أزواج "N".
import haiku as hk
import jax
import jax . numpy as jnp
from nucleotide_transformer . pretrained import get_pretrained_segment_nt_model
# Initialize CPU as default JAX device. This makes the code robust to memory leakage on
# the devices.
jax . config . update ( "jax_platform_name" , "cpu" )
backend = "cpu"
devices = jax . devices ( backend )
num_devices = len ( devices )
print ( f"Devices found: { devices } " )
# The number of DNA tokens (excluding the CLS token prepended) needs to be dividible by
# 2 to the power of the number of downsampling block, i.e 4.
max_num_nucleotides = 8
assert max_num_nucleotides % 4 == 0 , (
"The number of DNA tokens (excluding the CLS token prepended) needs to be dividible by"
"2 to the power of the number of downsampling block, i.e 4." )
parameters , forward_fn , tokenizer , config = get_pretrained_segment_nt_model (
model_name = "segment_nt" ,
embeddings_layers_to_save = ( 29 ,),
attention_maps_to_save = (( 1 , 4 ), ( 7 , 10 )),
max_positions = max_num_nucleotides + 1 ,
)
forward_fn = hk . transform ( forward_fn )
apply_fn = jax . pmap ( forward_fn . apply , devices = devices , donate_argnums = ( 0 ,))
# Get data and tokenize it
sequences = [ "ATTCCGATTCCGATTCCAACGGATTATTCCGATTAACCGATTCCAATT" , "ATTTCTCTCTCTCTCTGAGATCGATGATTTCTCTCTCATCGAACTATG" ]
tokens_ids = [ b [ 1 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens_str = [ b [ 0 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens = jnp . asarray ( tokens_ids , dtype = jnp . int32 )
random_key = jax . random . PRNGKey ( seed = 0 )
keys = jax . device_put_replicated ( random_key , devices = devices )
parameters = jax . device_put_replicated ( parameters , devices = devices )
tokens = jax . device_put_replicated ( tokens , devices = devices )
# Infer on the sequence
outs = apply_fn ( parameters , keys , tokens )
# Obtain the logits over the genomic features
logits = outs [ "logits" ]
# Transform them in probabilities
probabilities = jnp . asarray ( jax . nn . softmax ( logits , axis = - 1 ))[..., - 1 ]
print ( f"Probabilities shape: { probabilities . shape } " )
print ( f"Features inferred: { config . features } " )
# Get probabilities associated with intron
idx_intron = config . features . index ( "intron" )
probabilities_intron = probabilities [..., idx_intron ]
print ( f"Intron probabilities shape: { probabilities_intron . shape } " )أسماء النماذج المدعومة هي:
يعمل الكود على GPU وTPU بفضل Jax!
يتم تدريب النماذج على تسلسلات يصل طولها إلى 1000 رمز مميز، بما في ذلك رمز <CLS> المُلحق تلقائيًا ببداية التسلسل. يبدأ برنامج الرمز المميز في عملية الترميز من اليسار إلى اليمين من خلال تجميع الأحرف "A" و"C" و"G" و"T" في 6 وحدات. تم اختيار الحرف "N" بحيث لا يتم تجميعه داخل k-mers، لذلك عندما يواجه المُرمز الحرف "N"، أو إذا كان عدد النيوكليوتيدات في التسلسل ليس مضاعفًا للـ 6، فسوف يقوم برمز النيوكليوتيدات دون تجميع هم. الأمثلة مذكورة أدناه:
dna_sequence_1 = "ACGTGTACGTGCACGGACGACTAGTCAGCA"
tokenized_dna_sequence_1 = [ < CLS > , < ACGTGT > , < ACGTGC > , < ACGGAC > , < GACTAG > , < TCAGCA > ]
dna_sequence_2 = "ACGTGTACNTGCACGGANCGACTAGTCTGA"
tokenized_dna_sequence_2 = [ < CLS > , < ACGTGT > , < A > , < C > , < N > , < TGCACG > , < G > , < A > , < N > , < CGACTA > , < GTCTGA > ]وبالتالي يمكن لجميع المحولات v1 وv2 أن تأخذ تسلسلات تصل إلى 5994 و12282 نيوكليوتيدات على التوالي إذا لم يكن هناك "N" بالداخل.
مجموعة النماذج المعروضة في هذا المستودع متاحة على مساحات الوجه المعانقة في Instadeep هنا: مساحة nucleotide transformer ومساحة nucleotide transformer الزراعية!
نشكر ماسا رولر، وكذلك أعضاء Rostlab، وخاصة توبياس أوليني، وإيفان كولوداروف، وبوركهارد روست على المناقشات البناءة التي ساعدت في تحديد اتجاهات البحث المثيرة للاهتمام. علاوة على ذلك، نعرب عن امتناننا لجميع أولئك الذين يودعون البيانات التجريبية في قواعد البيانات العامة، وأولئك الذين يحافظون على قواعد البيانات هذه، وأولئك الذين يجعلون الأساليب التحليلية والتنبؤية متاحة مجانًا. ونشكر أيضًا فريق تطوير Jax.
إذا وجدت هذا المستودع مفيدًا في عملك، فيرجى إضافة اقتباس ذي صلة إلى أي من أوراقنا المرتبطة:
ورقة nucleotide transformer :
@article { dalla2023nucleotide ,
title = { The nucleotide transformer : Building and Evaluating Robust Foundation Models for Human Genomics } ,
author = { Dalla-Torre, Hugo and Gonzalez, Liam and Mendoza Revilla, Javier and Lopez Carranza, Nicolas and Henryk Grywaczewski, Adam and Oteri, Francesco and Dallago, Christian and Trop, Evan and Sirelkhatim, Hassan and Richard, Guillaume and others } ,
journal = { bioRxiv } ,
pages = { 2023--01 } ,
year = { 2023 } ,
publisher = { Cold Spring Harbor Laboratory }
}ورقة nucleotide transformer الزراعية:
@article { mendoza2024foundational ,
title = { A foundational large language model for edible plant genomes } ,
author = { Mendoza-Revilla, Javier and Trop, Evan and Gonzalez, Liam and Roller, Ma{v{s}}a and Dalla-Torre, Hugo and de Almeida, Bernardo P and Richard, Guillaume and Caton, Jonathan and Lopez Carranza, Nicolas and Skwark, Marcin and others } ,
journal = { Communications Biology } ,
volume = { 7 } ,
number = { 1 } ,
pages = { 835 } ,
year = { 2024 } ,
publisher = { Nature Publishing Group UK London }
}ورقة SegmentNT
@article { de2024segmentnt ,
title = { SegmentNT: annotating the genome at single-nucleotide resolution with DNA foundation models } ,
author = { de Almeida, Bernardo P and Dalla-Torre, Hugo and Richard, Guillaume and Blum, Christopher and Hexemer, Lorenz and Gelard, Maxence and Pandey, Priyanka and Laurent, Stefan and Laterre, Alexandre and Lang, Maren and others } ,
journal = { bioRxiv } ,
pages = { 2024--03 } ,
year = { 2024 } ,
publisher = { Cold Spring Harbor Laboratory }
}إذا كانت لديك أي أسئلة أو تعليقات حول الكود والنماذج، فلا تتردد في التواصل معنا.
شكرا لاهتمامك بعملنا!


