binnacle
1.0.0
ل
يحسب Binnacle بدقة تغطية سقالات الرسم البياني ويتكامل بسلاسة مع أساليب Binning الرائدة مثل Metabat2 و Maxbin 2.0 و Cofct. باستخدام سقالات الرسم البياني ، على عكس contigs (النهج الأكثر شيوعًا) لـ Binning ، يحسن تواصل ونوعية صناديق metagenomic ويمكن أن تلتقط مجموعة أوسع من عناصر التبعية للجينوم المعاد بناؤها.
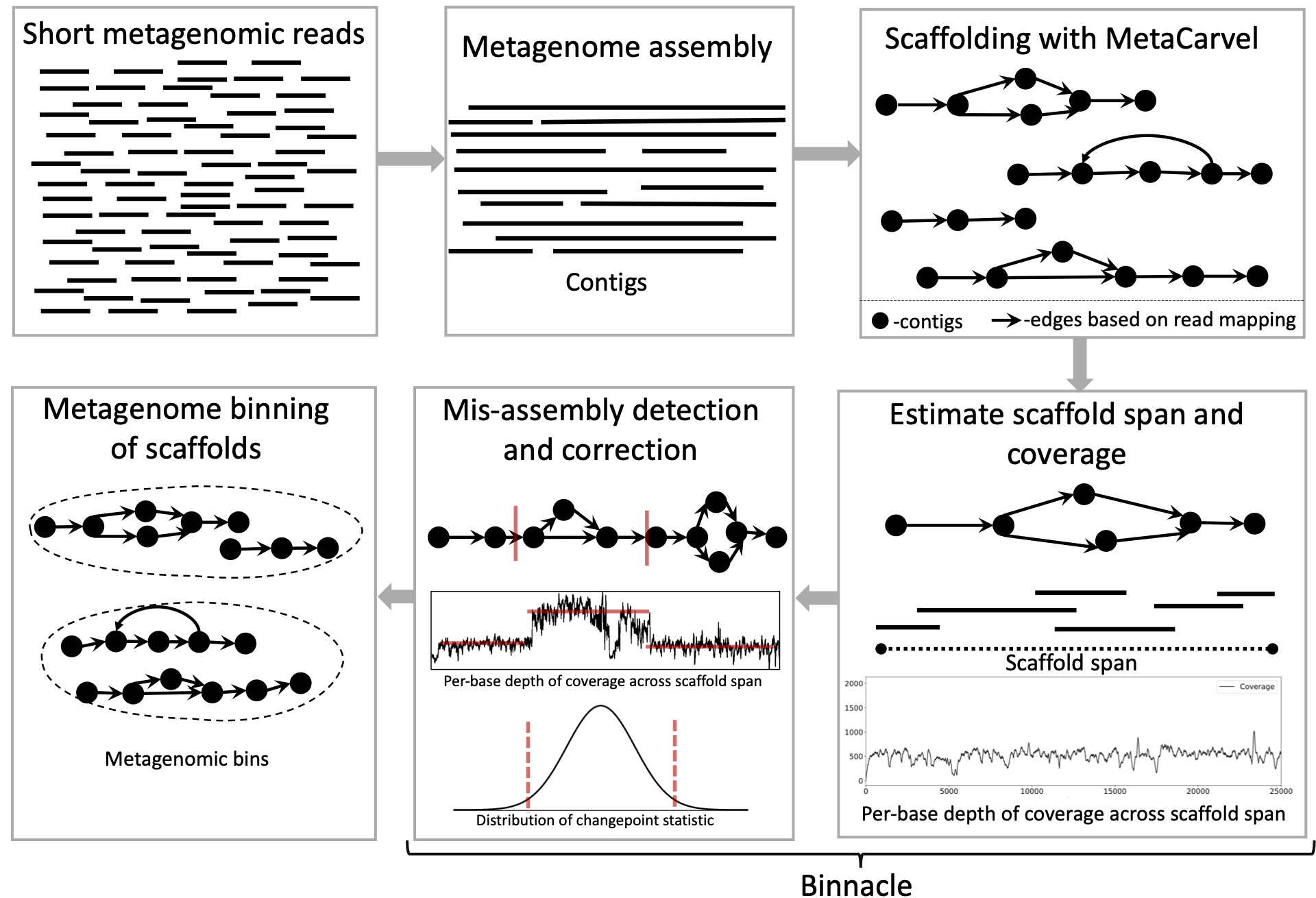
لتشغيل Binnacle ، ستحتاج إلى Python 3.7.x ، Bedtools ، Samtools ، Biopython ، NetworkX ، Numpy ، و Pandas.
يتوفر ملف البيئة. يتم تقديم الوثائق التفصيلية حول كيفية تثبيت هذه الحزم هنا. نستخدم سقالات الرسم البياني التي يتم إخراج أداة السقالات metacarvel ، لذلك ستحتاج أيضًا إلى تنزيل وتثبيت metacarvel. هناك دليل تثبيت خطوة بخطوة لـ Metacarvel.
بشكل عام ، عندما يكون لديك عينة أو عدة عينة من metagenomic ، نحتاج إلى تجميع السقالات/السقالات ، والسقالات من كل عينة لإنشاء صناديق metagenomic. نوصي باستخدام Megahit للتجميع ، و Metacarvel للسقالات. نحن نقدم دليلًا مساعد للعمل من خلال خطوات تقدير التجميع ، والسقالات ، وتقدير التغطية لكل قاعدة هنا.
اتبع هذه الخطوات لإنشاء ملفات لتشغيل أساليب binning مع سقالات الرسم البياني:
python Estimate_Abundances.py -g [ORIENTED.gml] -a [COVERAGE_SORTED.txt] -c [CONTIGS.fa] -d [OUTPUT_DIRECTORY]
usage: Estimate_Abundances.py [-h] [-g ASSEMBLY] [-a COVERAGE] [-bam BAMFILE]
[-bed BEDFILE] [-c CONTIGS] -d DIR [-o COORDS]
[-w WINDOW_SIZE] [-t THRESHOLD]
[-n NEIGHBOR_CUTOFF] [-p POSCUTOFF]
[-pre PREFIX]
binnacle: A tool for binning metagenomic datasets using assembly graphs and
scaffolds generated by metacarvel. Estimate_Abundances.py estimates abundance
for scaffolds generated by MetaCarvel. If the coordinates computed by binnacle
is specified then the abundance for each scaffold is estimated based on the
contig abundances and the coordinates. If the coordinates are not specified
then binnacle etimates the abundance from scratch. While calculating all vs
all abundances please specify the coordinates(Coordinates_After_Delinking.txt)
through the "coords" parameter. The abundances can be provided as a bed file,
bam file or a text file describing the per base coverage obtained by running
the genomeCoverageBed program of the bedtools suite.
optional arguments:
-h, --help show this help message and exit
-g ASSEMBLY, --assembly ASSEMBLY
Assembly Graph generated by Metacarvel
-a COVERAGE, --coverage COVERAGE
Output generated by running genomecov -d on the bed
file generated by MetaCarvel.
-bam BAMFILE, --bamfile BAMFILE
Bam file from aligning reads to contigs
-bed BEDFILE, --bedfile BEDFILE
Bed file from aligning reads to contigs. If bed file
is provided please provide a fasta file of the contigs
-c CONTIGS, --contigs CONTIGS
Contigs generated by the assembler, contigs.fasta
-d DIR, --dir DIR output directory for results
-o COORDS, --coords COORDS
Coordinate file generated by Binnacle
-w WINDOW_SIZE, --window_size WINDOW_SIZE
Size of the sliding window for computing test
statistic to identify changepoints in coverages
(Default=1500)
-t THRESHOLD, --threshold THRESHOLD
Threshold to identify outliers (Default=99)
-n NEIGHBOR_CUTOFF, --neighbor_cutoff NEIGHBOR_CUTOFF
Filter size to identify outliers within (Defualt=100)
-p POSCUTOFF, --poscutoff POSCUTOFF
Position cutoff to consider delinking (Default=100)
-pre PREFIX, --prefix PREFIX
Prefix to be attached to all outputs
-g Path to oriented.gml from running metacarvel on sample
-c Path to contigs obtained by assembling reads of sample
-a Coverage of contigs in ths sample by mapping to its reads -- See Wiki for how to calculate coverage information
-d Output directory
-a Coverage of contigs in Sample 1 by mapping reads of Sample 2 -- See Wiki for how to calculate coverage information
-o Coordinates of scaffolds from Sample 1 that you would have generated from the previous step.
-d Same output directory as Sample 1
python Collate.py -h
usage: Collate.py [-h] -d DIR [-m METHOD] [-k KEEP]
binnacle: A tool for binning metagenomic datasets using assembly graphs and
scaffolds generated by metacarvel.Estimate_Abundances.py estimates abundance
for scaffolds generated by MetaCarvel. The program Collate.py collects the
summary files generated by Estimate_Abundances.py
optional arguments:
-h, --help show this help message and exit
-d DIR, --dir DIR Output directory that contains the summary files
generated by running Estimate_Abundances.py
-m METHOD, --method METHOD
Binning method to format the output to. Presently we
support 1. Metabat 2. Maxbin 3. Concoct 4. Binnacle
(Default)
-k KEEP, --keep KEEP Retain the summary files generated by
Estimate_Abundances.py. Defaults to True
يرجى الخروج من الويكي للحصول على وصف مفصل عند إعداد بيئة Python ، وطرق حساب التغطية وسير عمل نموذجي لتشغيل Binnacle.
لتصور سقالات الرسم البياني ، نوصي باستخدام metagenomescope وهو متصفح يعتمد على الويب. المدخلات إلى metagenomescope هي Assembly_graph_filtered.gml. يتم تقديم وثائق مفصلة حول تثبيت وتشغيل metagenomescope هنا.
يرجى الاستشهاد بـ Muralidharan HS و Shah N و Meisel JS و Pop M (2021) Binnacle: باستخدام سقالات لتحسين تواصل وجودة صناديق الميتاجنومية. أمام. ميكروبول. 12: 638561. doi: 10.3389/fmicb.2021.638561.
هذه الأداة لا تزال قيد التطوير. يرجى فتح المشكلة هنا على github أو الاتصال بنا إذا كان لديك أي أسئلة.
Harihara Muralidharan: [email protected]
Nidhi Shah: [email protected]


