binnacle
1.0.0
Zu
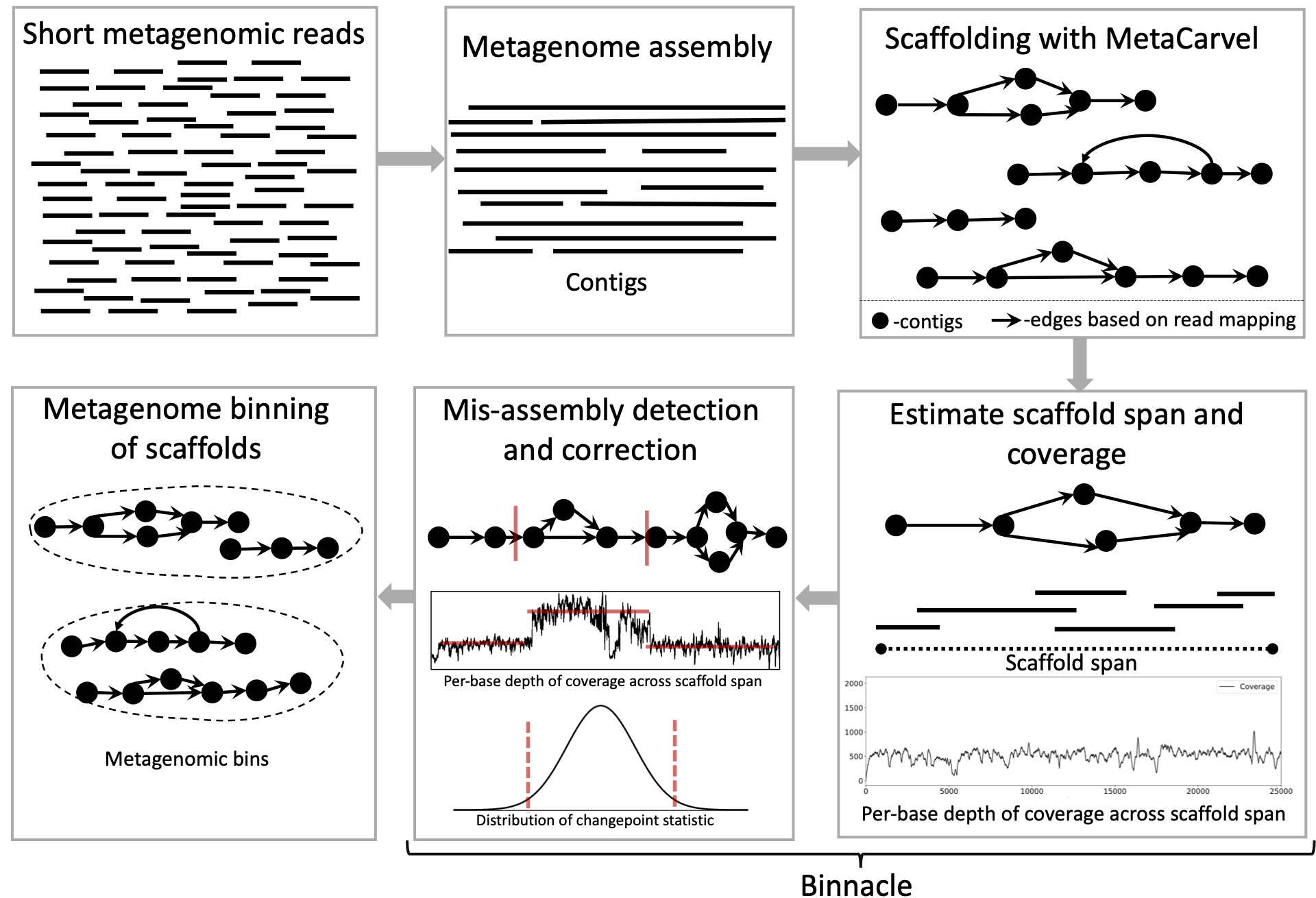
Binnacle berechnet die Abdeckung von Graph -Gerüsten genau und integriert sich nahtlos in führende Binning -Methoden wie Metabat2, Maxbin 2.0 und cococt. Die Verwendung von Graph -Gerüsten im Gegensatz zu Contigs (häufigsten Ansatz) verbessert die Kontiguität und Qualität der metagenomischen Mülleimer und kann einen breiteren Satz der akzessorischen Elemente der rekonstruierten Genome erfassen.
Um Binnacle zu leiten, benötigen Sie Python 3.7.x, Bedtools, Samtools, Biopython, Networkx, Numpy und Pandas.
Eine umwelt.yml -Datei ist verfügbar und kann verwendet werden, um eine Conda -Umgebung zu erstellen, die für den Ausführen von Binnacle geeignet ist. Die detaillierte Dokumentation zum Installieren dieser Pakete finden Sie hier. Wir verwenden Graph -Gerüste, die das Metacarvel -Gerüstwerkzeug ausgeben, sodass Sie auch Metacarvel herunterladen und installieren müssen. Es gibt einen Schritt -für -Schritt -Installationshandbuch für Metacarvel.
Wenn Sie eine oder mehrere metagenomische Proben haben, müssen wir im Allgemeinen zusammenstellen, Gerüst- und Behälter -Contigs/-gerüste aus jeder Probe montagen, um metagenomische Behälter zu erzeugen. Wir empfehlen, Megahit für die Montage und Metacarvel für Gerüste zu verwenden. Wir bieten hier einen Helfer-Leitfaden zur Arbeit durch die Schätzung der Versammlung, des Gerüsts und der Abdeckung pro Basis.
Befolgen Sie diese Schritte, um Dateien zum Ausführen von Binning -Methoden mit Grafikgeräten zu generieren:
python Estimate_Abundances.py -g [ORIENTED.gml] -a [COVERAGE_SORTED.txt] -c [CONTIGS.fa] -d [OUTPUT_DIRECTORY]
usage: Estimate_Abundances.py [-h] [-g ASSEMBLY] [-a COVERAGE] [-bam BAMFILE]
[-bed BEDFILE] [-c CONTIGS] -d DIR [-o COORDS]
[-w WINDOW_SIZE] [-t THRESHOLD]
[-n NEIGHBOR_CUTOFF] [-p POSCUTOFF]
[-pre PREFIX]
binnacle: A tool for binning metagenomic datasets using assembly graphs and
scaffolds generated by metacarvel. Estimate_Abundances.py estimates abundance
for scaffolds generated by MetaCarvel. If the coordinates computed by binnacle
is specified then the abundance for each scaffold is estimated based on the
contig abundances and the coordinates. If the coordinates are not specified
then binnacle etimates the abundance from scratch. While calculating all vs
all abundances please specify the coordinates(Coordinates_After_Delinking.txt)
through the "coords" parameter. The abundances can be provided as a bed file,
bam file or a text file describing the per base coverage obtained by running
the genomeCoverageBed program of the bedtools suite.
optional arguments:
-h, --help show this help message and exit
-g ASSEMBLY, --assembly ASSEMBLY
Assembly Graph generated by Metacarvel
-a COVERAGE, --coverage COVERAGE
Output generated by running genomecov -d on the bed
file generated by MetaCarvel.
-bam BAMFILE, --bamfile BAMFILE
Bam file from aligning reads to contigs
-bed BEDFILE, --bedfile BEDFILE
Bed file from aligning reads to contigs. If bed file
is provided please provide a fasta file of the contigs
-c CONTIGS, --contigs CONTIGS
Contigs generated by the assembler, contigs.fasta
-d DIR, --dir DIR output directory for results
-o COORDS, --coords COORDS
Coordinate file generated by Binnacle
-w WINDOW_SIZE, --window_size WINDOW_SIZE
Size of the sliding window for computing test
statistic to identify changepoints in coverages
(Default=1500)
-t THRESHOLD, --threshold THRESHOLD
Threshold to identify outliers (Default=99)
-n NEIGHBOR_CUTOFF, --neighbor_cutoff NEIGHBOR_CUTOFF
Filter size to identify outliers within (Defualt=100)
-p POSCUTOFF, --poscutoff POSCUTOFF
Position cutoff to consider delinking (Default=100)
-pre PREFIX, --prefix PREFIX
Prefix to be attached to all outputs
-g Path to oriented.gml from running metacarvel on sample
-c Path to contigs obtained by assembling reads of sample
-a Coverage of contigs in ths sample by mapping to its reads -- See Wiki for how to calculate coverage information
-d Output directory
-a Coverage of contigs in Sample 1 by mapping reads of Sample 2 -- See Wiki for how to calculate coverage information
-o Coordinates of scaffolds from Sample 1 that you would have generated from the previous step.
-d Same output directory as Sample 1
python Collate.py -h
usage: Collate.py [-h] -d DIR [-m METHOD] [-k KEEP]
binnacle: A tool for binning metagenomic datasets using assembly graphs and
scaffolds generated by metacarvel.Estimate_Abundances.py estimates abundance
for scaffolds generated by MetaCarvel. The program Collate.py collects the
summary files generated by Estimate_Abundances.py
optional arguments:
-h, --help show this help message and exit
-d DIR, --dir DIR Output directory that contains the summary files
generated by running Estimate_Abundances.py
-m METHOD, --method METHOD
Binning method to format the output to. Presently we
support 1. Metabat 2. Maxbin 3. Concoct 4. Binnacle
(Default)
-k KEEP, --keep KEEP Retain the summary files generated by
Estimate_Abundances.py. Defaults to True
Bitte überprüfen Sie das Wiki für eine detaillierte Beschreibung zum Einrichten der Python -Umgebung, Methoden zur Berechnung der Abdeckung und eines typischen Workflows, um Binnacle auszuführen.
Um die Graph-Gerüste zu visualisieren, empfehlen wir die Verwendung von Metagenomescope, einem webbasierten Browser. Die Eingabe in Metagenomescope ist Assembly_Graph_Filtered.gml. Eine detaillierte Dokumentation zum Installieren und Ausführen von Metagenomescope finden Sie hier.
Bitte zitieren Sie Muralidharan HS, Shah N, Meisel JS und Pop M (2021) Binnacle: Verwenden von Gerüsten zur Verbesserung der Kontiguität und Qualität der metagenomischen Mülleimer. Front. Mikrobiol. 12: 638561. doi: 10.3389/fmicb.2021.638561.
Dieses Tool befindet sich noch in der Entwicklung. Bitte öffnen Sie hier auf GitHub das Problem oder kontaktieren Sie uns, wenn Sie Fragen haben.
Harihara Muralidharan: [email protected]
Nidhi Shah: [email protected]


