CELL E_2
1.0.0

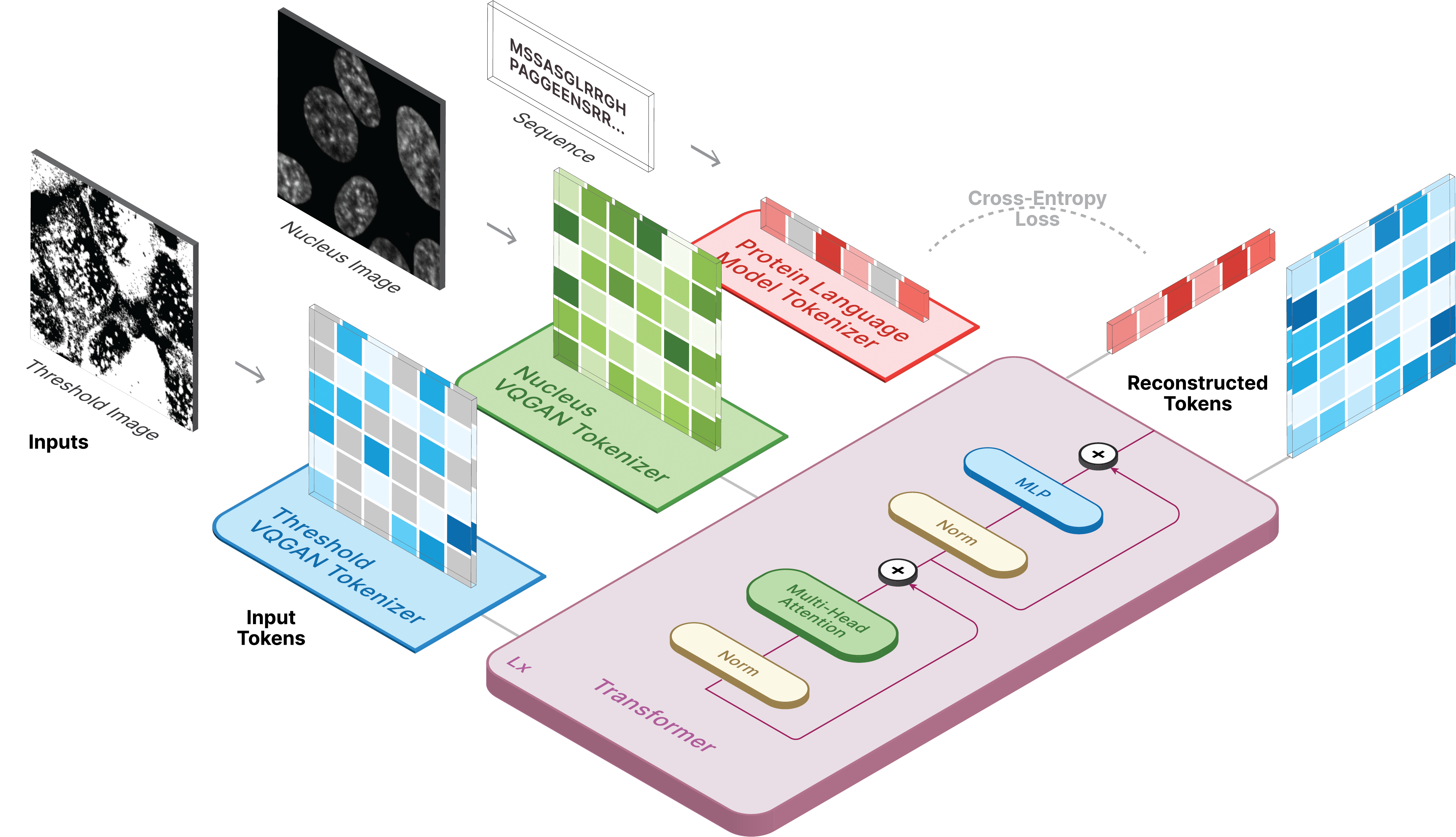
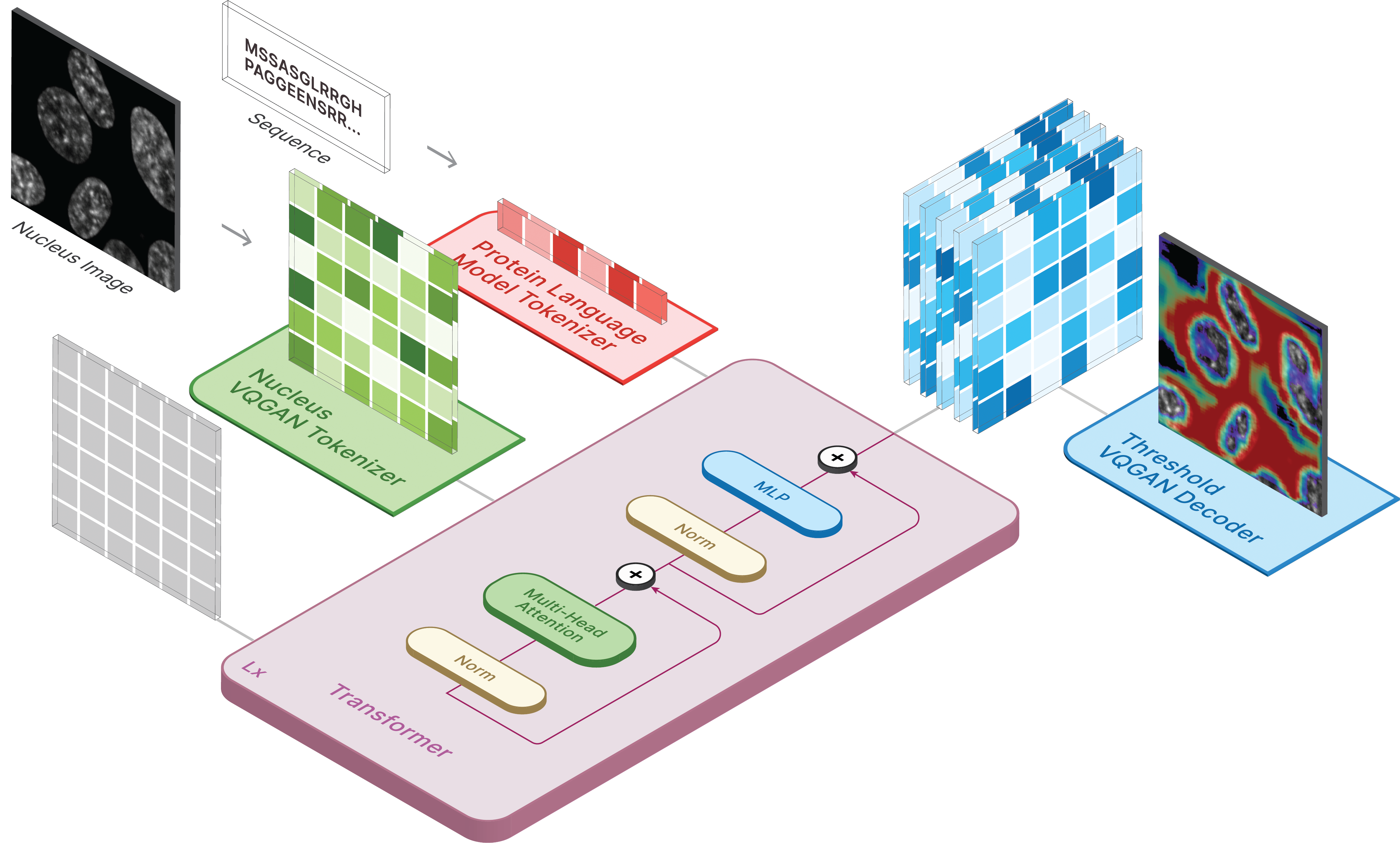
This repository is the official implementation of CELL-E 2: Translating Proteins to Pictures and Back with a Bidirectional Text-to-Image Transformer.
Create a virtual environment and install the required packages via:
pip install -r requirements.txt
Next, install torch = 2.0.0 with the appropriate CUDA version
Models are available on Hugging Face.
We also have two spaces available where you can run predictions on your own data!
To generate images, set the saved model as the ckpt_path. This method can be unstable, so refer to Demo.ipynb to see another way of loading.
from omegaconf import OmegaConf
from celle_main import instantiate_from_config
configs = OmegaConf.load(configs/celle.yaml);
model = instantiate_from_config(configs.model).to(device);
model.sample(text=sequence,
condition=nucleus,
return_logits=True,
progress=True)model.sample_text(condition=nucleus,
image=image,
return_logits=True,
progress=True)Training for CELL-E occurs in 3 stages:
If using the protein threshold image, set threshold: True for the dataset.
We use a slightly modified version of the taming-transformers code.
To train, run the following script:
python celle_taming_main.py --base configs/threshold_vqgan.yaml -t True
Please refer to the original repo for additional flags, such as --gpus.
We provide scripts for downloading Human Protein Atlas and OpenCell images in the scripts folder. A data_csv is needed to for the dataloader. You must generate a csv file which contains the columns nucleus_image_path, protein_image_path, metadata_path, split (train or val), and sequence (optional). It is assumed that this file exists within the the same general data folder as the images and metadata files.
Metadata is a JSON which should accompany every protein sequence. If a sequence does not appear in the data_csv, it must appear in metadata.json with the a key named protein_sequence.
Adding more information here can be useful for querying individual proteins. They can be retrieved via retrieve_metadata, which creates a self.metadata variable within the dataset object.
To train, run the following script:
python celle_main.py --base configs/celle.yaml -t True
Specify --gpus in the same format as VQGAN.
CELL-E contains the following options:
ckpt_path : Resume previous CELL-E 2 training. Saved model with state_dictvqgan_model_path : Saved protein image model (with state_dict) for protein image encodervqgan_config_path: Saved protein image model yamlcondition_model_path : Saved condition (nucleus) model (with state_dict) for protein image encodercondition_config_path: Saved condition (nucleus) model yamlnum_images: 1 if only using protein image encoder, 2 if including condition image encoderimage_key: nucleus, target, or threshold
dim: Dimension of language model embeddingnum_text_tokens: total number of tokens in language model (33 for ESM-2)text_seq_len: Total number of amino acids considereddepth: Transformer model depth, deeper is usually better at the cost of VRAMheads: number of heads used in multi-headed attentiondim_head: size of attention headsattn_dropout: Attention Dropout rate in trainingff_dropout: Feed-Forward Dropout rate in trainingloss_img_weight: Weighting applied to image reconstruction. text weight = 1loss_text_weight: Weighting applied to condition image reconstruction.stable: Norms weights (for when exploding gradients occur)learning_rate: Learning rate for Adam optimizermonitor: Param used to save modelsPlease cite us if you decide to use our code for any part of your research.
@inproceedings{
anonymous2023translating,
title={CELL-E 2: Translating Proteins to Pictures and Back with a Bidirectional Text-to-Image Transformer},
author={Emaad Khwaja, Yun S. Song, Aaron Agarunov, and Bo Huang},
booktitle={Thirty-seventh Conference on Neural Information Processing Systems},
year={2023},
url={https://openreview.net/forum?id=YSMLVffl5u}
}


