ClockstaR
1.0.0

Sebastian Duchene, Martyna Molak y Simon YW Ho.
Laboratorio de Ecología Molecular, Evolución y Filogenética (MEEP)
Facultad de Ciencias Biológicas
Universidad de Sídney
10 junio, 2015
partition_data_partitionfinder('drag fasta file with concatenated data here', 'drag partition finder output here')
optim.trees.interactive(folder.parts = 'path to your folder with fasta files and tree topology here')
Implementar la optimización de las distancias de los árboles utilizando la derivada de la distancia BSD.
Implementar la versión paralela para la distancia de topología.
Tutorial de escritura para agrupación a distancia de topología
Integre el generador de modelos para pruebas de modelos
Integre RaxML para una optimización de máxima probabilidad de longitudes y topologías de sucursales
Estimar escalas de tiempo evolutivas con conjuntos de datos multigénicos es un ejercicio común en los estudios filogenéticos. Los conjuntos de datos multigénicos se pueden dividir por gen, posición de codón o ambos. En este tutorial, nos referimos a "subconjuntos de datos" como genes individuales o cualquier subunidad del conjunto de datos multigénico. El término "particiones" se referirá a un grupo de subconjuntos de datos.
Aunque los subconjuntos de datos se pueden concatenar y analizar con un único modelo de reloj relajado, los patrones de variación de la tasa entre linajes pueden diferir entre los subconjuntos de datos incluso cuando sus topologías de árbol son idénticas. Por ejemplo, la tasa de variación entre linajes en los genes mitocondriales puede diferir de la de los genes nucleares. Por lo tanto, se pueden asignar diferentes modelos de reloj relajado a diferentes subconjuntos de datos para mejorar las estimaciones de las escalas de tiempo evolutivas y el ajuste estadístico (ver Duchene y Ho., 2014a).
Hay una gran cantidad de formas en que se pueden dividir conjuntos de datos multigénicos. Un enfoque común para comparar esquemas de partición es utilizar factores Bayes o criterios basados en probabilidad para el ajuste del modelo. Sin embargo, en la mayoría de los casos, no es factible probar todos los esquemas de partición posibles, especialmente con métodos computacionales intensivos para calcular factores de Bayes.
ClockstaR estima las longitudes de las ramas filogenéticas de cada subconjunto de datos. La distancia de puntuación de rama, conocida como sBSDmin, se calcula para cada par de árboles como una medida de la diferencia en sus patrones de variación de la tasa entre linajes. Estas distancias se utilizan para inferir la mejor estrategia de partición utilizando la estadística GAP con el algoritmo de agrupamiento PAM, tal como se implementa en el grupo de paquetes (Maechler et al., 2012) (para obtener detalles de la métrica sBSDmin, consulte Duchene et al., 2014b). .
ClockstaR es un paquete R para análisis filogenéticos de relojes moleculares de conjuntos de datos de múltiples genes. Utiliza los patrones de variación de la tasa entre linajes para los diferentes genes para seleccionar la estrategia de partición del reloj. El método utiliza una métrica de distancia de árbol filogenético y un algoritmo de aprendizaje automático no supervisado para identificar el número óptimo de particiones de reloj y qué genes deben analizarse en cada una de las particiones. La estrategia de partición seleccionada en ClocsktaR se puede utilizar para análisis posteriores del reloj molecular con programas como BEAST, MrBayes, PhyloBayes y otros.
Siga este enlace para ver la publicación original.
ClockstaR requiere una instalación de R. También requiere algunas dependencias de R, que se pueden obtener a través de R, como se explica a continuación.
Envíe cualquier solicitud o pregunta a Sebastian Duchene (sebastian.duchene[at]sydney.edu.au). Se pueden encontrar otros programas y recursos en el Laboratorio de Ecología Molecular, Evolución y Filogenética de la Universidad de Sydney.
Descargue este repositorio como un archivo zip y descomprímalo. Las siguientes instrucciones utilizan la carpeta clockstar_example_data, que contiene algunos archivos fasta y un árbol filogenético en formato newick. Abra cualquiera de estos archivos en un editor de texto, como Text Wrangler. Estos datos fueron simulados bajo cuatro patrones de variación de la tasa evolutiva. Tenga en cuenta que el árbol es la topología de árbol para todos los genes o particiones de datos. Para ejecutar ClockstaR, formatee sus datos de manera similar a los datos de ejemplo en clockstar_example_data.
ClockstaR se puede instalar directamente desde GitHub. Esto requiere el paquete devtools. Escriba el siguiente código en el indicador de R para instalar todas las herramientas necesarias (tenga en cuenta que necesitará conexión a Internet para descargar los paquetes directamente):
install . packages ( " devtools " )
library (devtools)
install_github ( ' ClockstaR ' , ' sebastianduchene ' )Después de descargar e instalar, cargue ClockstaR con la biblioteca de funciones.
library (ClockstaR2)Para ver un ejemplo de cómo se ejecuta el programa escriba:
example (ClockstaR2)El resto de este tutorial utiliza la carpeta clockstar_example_data
El primer paso es obtener los árboles genéticos de cada uno de los alineamientos. Para hacer esto, utilizamos una topología de árbol y optimizamos las longitudes de las ramas usando cada una de las alineaciones de genes individuales, en este caso desde A1.fasta hasta C3.fasta. Si tiene los árboles genéticos, guárdelos en formato newick en un archivo y vaya al siguiente paso (ejecutar clockstar de forma interactiva).
Escriba el siguiente código en el indicador de R y presione Intro:
optim . trees . interactive ()Si recibe un mensaje de error sobre la instalación del paquete phangorn, utilice este código y luego repita optim.trees.interactive()
install . packcages ( " phangorn " )ClockstaR imprimirá el siguiente mensaje:
Please drag a folder with the data subsets and a tree topology . The files should be in FASTA format, and the trees in NEWICKArrastre la carpeta clockstar_example_data a la consola R y escriba enter. Tenga en cuenta que la carpeta solo debe contener las alineaciones en formato FASTA y la topología del árbol en NEWICK. Verá el siguiente mensaje:
What should be the name of the file to save the optimised trees ?Escriba el nombre del archivo para los árboles optimizados. En este caso usaremos "example.trees"
example . treesEn este punto, ClockstaR preguntará si debería utilizar un modelo de sustitución independiente para cada gen o utilizar JC en todos los casos. Dado que estos datos fueron simulados bajo JC, escribiremos "n" y presionaremos Intro. Escriba "y" para especificar cada modelo de sustitución por separado.
Después de escribir "n" y presionar Enter, ClockstaR comenzará a ejecutarse. Imprimirá los árboles genéticos en el dispositivo gráfico. Si el árbol especificado estaba rooteado, también puede imprimir algunas advertencias, que pueden ignorarse con seguridad.
Abra la carpeta clockstar_example_data. Encontrará un archivo con el nombre "example.trees", como se especifica unos pasos más arriba. Abra example.trees en un editor de texto. Contiene cada árbol de genes y los nombres de los árboles, de acuerdo con los nombres de las alineaciones de genes. Debería verse así:
A1 . fasta (( t1 : 0.01504695462 ,( t2 : 0.00987 ...
A2 . fasta (( t1 : 0.01520523401 ,( t2 : 0.01317 ...
A3 . fasta (( t1 : 0.01519309467 ,( t2 : 0.01092 ...
.
.
.Este archivo con árboles se utilizará para el siguiente paso.
Para este paso es necesario tener los árboles de genes en un archivo, como el obtenido en el paso anterior.
Abra R y cargue ClockstaR como se muestra arriba. Escriba el siguiente código cuando se le solicite:
clockstar . interactive ()ClockstaR imprimirá el siguiente mensaje:
please drag or type in the path to your gene trees file in NEWICK format :Arrastra el archivo con los árboles genéticos a la consola R. Si siguió el paso anterior, el archivo se llamará ejemplo.trees. Escribe enter.
Dependiendo de los paquetes que haya instalado, ClockstaR puede preguntar si debe ejecutarse en paralelo. Esto es eficaz para grandes conjuntos de datos. Pero para los datos de ejemplo no habrá una gran diferencia, así que escriba "n" si ve este mensaje y luego escriba enter:
Packages foreach and doParallel are available for parallel computation
Should we run ClockstaR in parallel (y / n) ? (This is good for large data sets)Clockstar ahora comenzará a funcionar. La salida en pantalla debería verse así:
[ 1 ] " Calculating sBSDmin distances between all pairs of trees "
[ 1 ] " Estimating tree distances "
[ 1 ] " estimating distances 1 of 11 "
[ 1 ] " estimating distances 2 of 11 "
[ 1 ] " estimating distances 3 of 11 "
[ 1 ] " estimating distances 4 of 11 "
[ 1 ] " estimating distances 5 of 11 "
.
.
.Después de estimar las distancias de los árboles (descritas en la publicación original), ClockstaR imprimirá el siguiente mensaje:
" I finished calculating the sBSDmin distances between trees "
The settings for clustering with ClockstaR are :
PAM clustering algorithm
K from 1 to number of data subsets - 1
SEmax criterion to select the optimal k
500 bootstrap replicates
Are these correct ? (y / n)Estas son las configuraciones para el algoritmo de agrupamiento. Son apropiados para la mayoría de los conjuntos de datos, por lo que en este ejemplo podemos escribir "y" y luego ingresar. Al escribir "n" podemos cambiar esta configuración; para obtener más detalles, consulte Kaufman y Rousseeuw (2009).
ClockstaR ahora ejecutará el algoritmo de agrupación. Al final, imprimirá la mejor cantidad de particiones y preguntará si los resultados deben guardarse en un archivo pdf:
[ 1 ] " ClockstaR has finished running "
[ 1 ] " The best number of partitions for your data set is: 3 "
Do you wish to save the results in a pdf file ? (y / n)Escriba "y" y luego ingrese.
ClockstaR luego le pedirá el nombre de los archivos de salida:
What should be the name and path of the output file ?Para este ejemplo, escriba "example_run" e ingrese, pero se puede usar cualquier nombre.
Ahora abra la carpeta clockstar_example_data y abra los dos archivos pdf, example_run_gapstats.pdf y example_run_matrix.pdf.
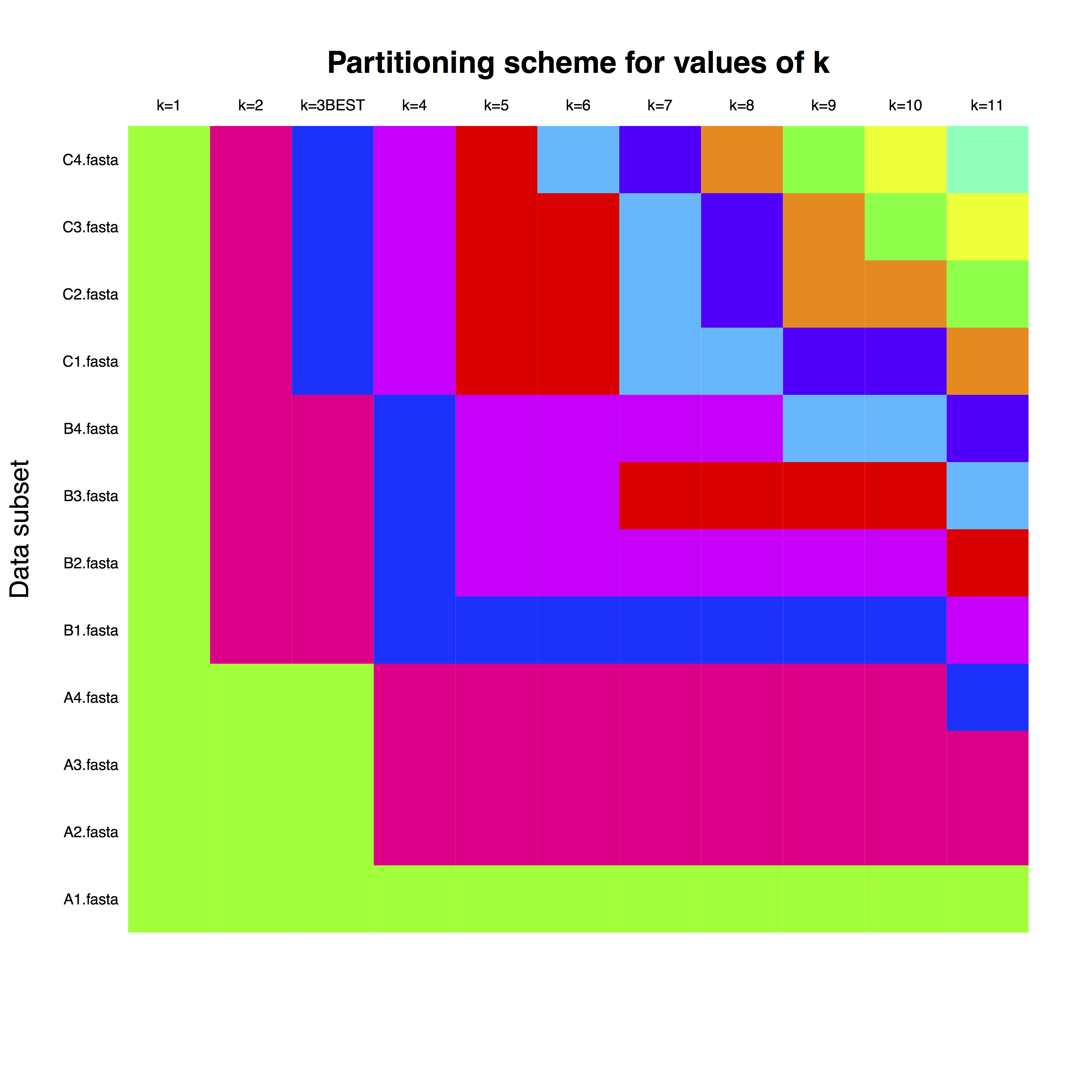
example_run_matrix es una matriz, donde las filas corresponden a cada gen, como se nombra en los archivos FASTA. Las columnas son el número de particiones y los colores representan la asignación de cada gen a la partición del reloj. Por ejemplo, para k = 3, que es el mejor número de particiones, se pueden usar particiones de reloj separadas para los genes con las letras A, B y C.
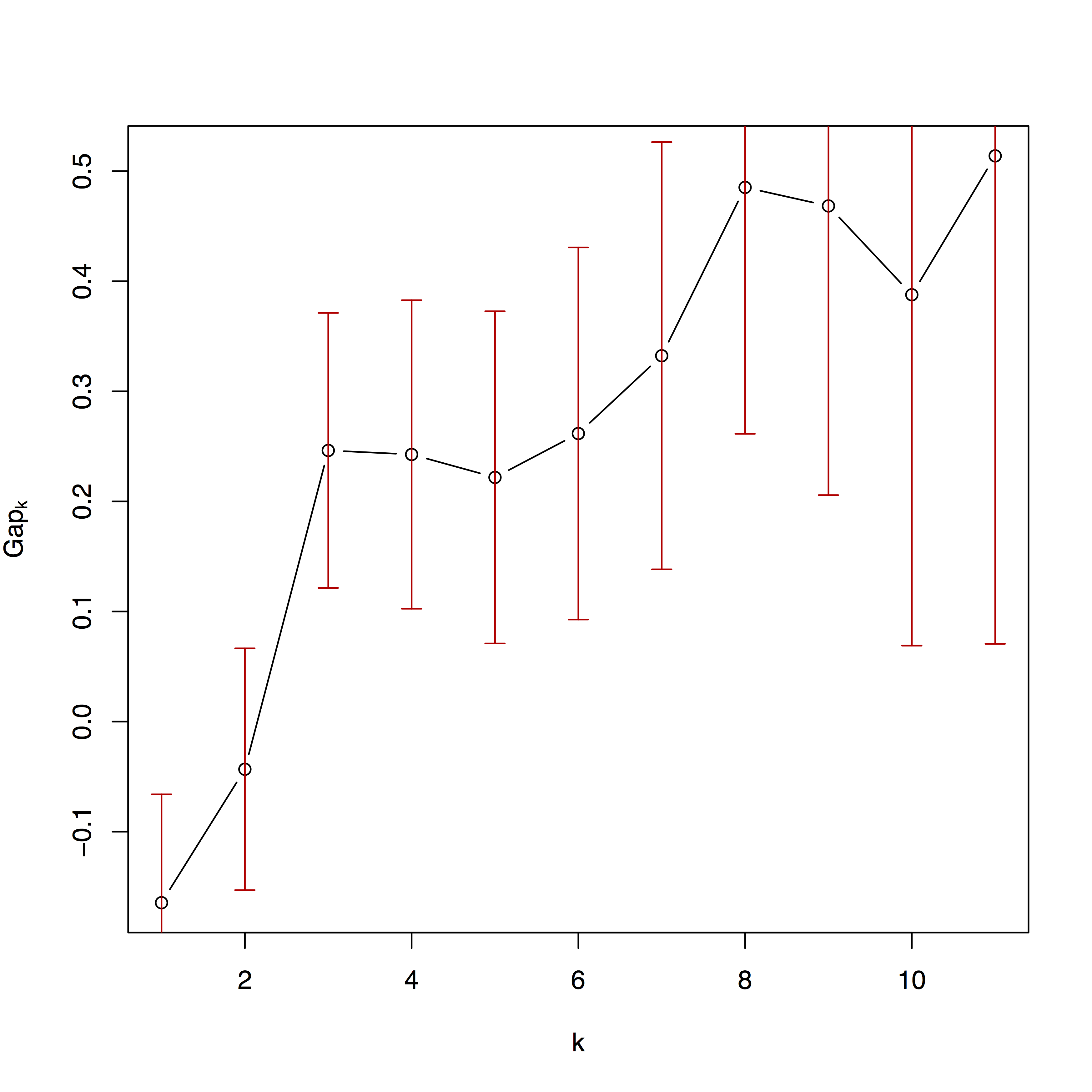
El segundo gráfico es el ajuste de los algoritmos de agrupamiento en diferentes números de particiones. Hay más detalles disponibles en Kaufman y Rousseeuw (2009) y en la documentación del grupo de paquetes.
ClockstaR se puede ejecutar con otras configuraciones personalizadas. Consulte la documentación para obtener más detalles o escríbame si tiene alguna pregunta a sebastian.duchene[at]sydney.edy.au.
El logo fue diseñado por Jun Tong.
Duchene, S. y Ho, SY (2014a). Uso de múltiples modelos de reloj relajado para estimar escalas de tiempo evolutivas a partir de datos de secuencias de ADN. Filogenética molecular y evolución (77): 65-70.
Duchene, S., Molak, M. y Ho, SY (2014b). ClockstaR: elección del número de modelos de reloj relajado en análisis filogenético molecular. Bioinformática 30 (7): 1017-1019.
Kaufman, L. y Rousseeuw, PJ (2009). Encontrar grupos en datos: una introducción al análisis de conglomerados (Vol. 344). John Wiley e hijos.