BindCraft
v1.2.0
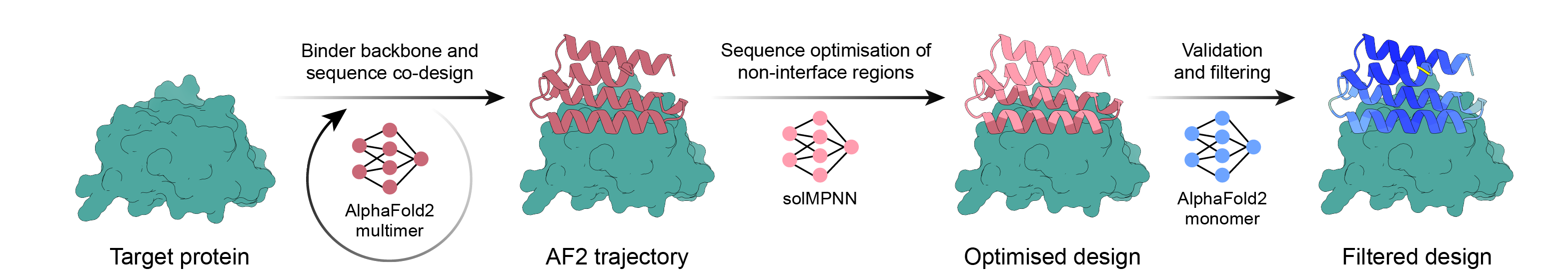
Pipeline de conception de liant simple utilisant la rétropropagation AlphaFold2, MPNN et PyRosetta. Sélectionnez votre cible et laissez le script faire le reste du travail et terminez une fois que vous avez suffisamment de designs à commander !
Lien de préimpression pour BindCraft
Vous devez d'abord cloner ce référentiel. Remplacez [install_folder] par le chemin où vous souhaitez l'installer.
git clone https://github.com/martinpacesa/BindCraft [install_folder]
Accédez à votre dossier d’installation à l’aide du CD et exécutez le code d’installation. BindCraft nécessite une carte graphique Nvidia compatible CUDA pour fonctionner. Dans le paramètre cuda , veuillez spécifier la version CUDA compatible avec votre carte graphique, par exemple '11.8'. En cas de doute, laissez ce champ vide, mais il est possible que l'installation sélectionne la mauvaise version, ce qui entraînera des erreurs. Dans pkg_manager, spécifiez si vous utilisez « mamba » ou « conda », si laissé vide, il utilisera « conda » par défaut.
Remarque : ce script d'installation installera PyRosetta, qui nécessite une licence à des fins commerciales.
bash install_bindcraft.sh --cuda '12.4' --pkg_manager 'conda'
Essayez toujours de réduire le PDB cible d'entrée à la plus petite taille possible ! Cela accélérera considérablement la génération du classeur et minimisera les besoins en mémoire GPU.
Soyez prêt à parcourir au moins quelques centaines de trajectoires pour voir certains classeurs acceptés, pour les cibles difficiles, cela peut même être quelques milliers.
Pour exécuter le script localement, vous devez d'abord configurer votre fichier .json cible dans le dossier settings_target . Dans le fichier json se trouvent les paramètres suivants :
design_path -> path where to save designs and statistics
binder_name -> what to prefix your designed binder files with
starting_pdb -> the path to the PDB of your target protein
chains -> which chains to target in your protein, rest will be ignored
target_hotspot_residues -> which position to target for binder design, for example `1,2-10` or chain specific `A1-10,B1-20` or entire chains `A`, set to null if you want AF2 to select binding site; better to select multiple target residues or a small patch to reduce search space for binder
lengths -> range of binder lengths to design
number_of_final_designs -> how many designs that pass all filters to aim for, script will stop if this many are reached
Exécutez ensuite le script de conception du classeur :
sbatch ./bindcraft.slurm --settings './settings_target/PDL1.json' --filters './settings_filters/default_filters.json' --advanced './settings_advanced/default_4stage_multimer.json'
L'indicateur de paramètres doit pointer vers votre .json cible que vous avez défini ci-dessus. L'indicateur de filtres pointe vers le json où les filtres de conception sont spécifiés (la valeur par défaut est ./filters/default_filters.json). L'indicateur avancé pointe vers vos paramètres avancés (la valeur par défaut est ./advanced_settings/default_4stage_multimer.json). Si vous omettez les filtres et les paramètres avancés, les paramètres par défaut seront automatiquement pointés.
Alternativement, si votre machine ne prend pas en charge SLURM, vous pouvez exécuter le code directement en activant l'environnement dans conda et en exécutant le code python :
conda activate BindCraft
cd /path/to/bindcraft/folder/
python -u ./bindcraft.py --settings './settings_target/PDL1.json' --filters './settings_filters/default_filters.json' --advanced './settings_advanced/default_4stage_multimer.json'
Nous recommandons de générer au moins 100 conceptions finales passant tous les filtres, puis de commander les 5 à 20 meilleures pour la caractérisation expérimentale. Si des liants à haute affinité sont nécessaires, il est préférable d'effectuer un dépistage davantage, car la métrique ipTM utilisée pour le classement n'est pas un bon prédicteur d'affinité, mais s'est avérée être un bon prédicteur binaire de liaison.
Vous trouverez ci-dessous des explications sur les filtres individuels et les paramètres avancés.
Voici les paramètres avancés contrôlant le processus de conception :
omit_AAs -> which amino acids to exclude from design (note: they can still occur if no other options are possible in the position)
force_reject_AA -> whether to force reject design if it contains any amino acids specified in omit_AAs
design_algorithm -> which design algorithm for the trajecory to use, the currently implemented algorithms are below
use_multimer_design -> whether to use AF2-ptm or AF2-multimer for binder design; the other model will be used for validation then
num_recycles_design -> how many recycles of AF2 for design
num_recycles_validation -> how many recycles of AF2 use for structure prediction and validation
sample_models = True -> whether to randomly sample parameters from AF2 models, recommended to avoid overfitting
rm_template_seq_design -> remove target template sequence for design (increases target flexibility)
rm_template_seq_predict -> remove target template sequence for reprediction (increases target flexibility)
rm_template_sc_design -> remove sidechains from target template for design
rm_template_sc_predict -> remove sidechains from target template for reprediction
# Design iterations
soft_iterations -> number of soft iterations (all amino acids considered at all positions)
temporary_iterations -> number of temporary iterations (softmax, most probable amino acids considered at all positions)
hard_iterations -> number of hard iterations (one hot encoding, single amino acids considered at all positions)
greedy_iterations -> number of iterations to sample random mutations from PSSM that reduce loss
greedy_percentage -> What percentage of protein length to mutate during each greedy iteration
# Design weights, higher value puts more weight on optimising the parameter.
weights_plddt -> Design weight - pLDDT of designed chain
weights_pae_intra -> Design weight - PAE within designed chain
weights_pae_inter -> Design weight - PAE between chains
weights_con_intra -> Design weight - maximise number of contacts within designed chain
weights_con_inter -> Design weight - maximise number of contacts between chains
intra_contact_distance -> Cbeta-Cbeta cutoff distance for contacts within the binder
inter_contact_distance -> Cbeta-Cbeta cutoff distance for contacts between binder and target
intra_contact_number -> how many contacts each contact esidue should make within a chain, excluding immediate neighbours
inter_contact_number -> how many contacts each contact residue should make between chains
weights_helicity -> Design weight - helix propensity of the design, Default 0, negative values bias towards beta sheets
random_helicity -> whether to randomly sample helicity weights for trajectories, from -1 to 1
# Additional losses
use_i_ptm_loss -> Use i_ptm loss to optimise for interface pTM score?
weights_iptm -> Design weight - i_ptm between chains
use_rg_loss -> use radius of gyration loss?
weights_rg -> Design weight - radius of gyration weight for binder
use_termini_distance_loss -> Try to minimise distance between N- and C-terminus of binder? Helpful for grafting
weights_termini_loss -> Design weight - N- and C-terminus distance minimisation weight of binder
# MPNN settings
mpnn_fix_interface -> whether to fix the interface designed in the starting trajectory
num_seqs -> number of MPNN generated sequences to sample and predict per binder
max_mpnn_sequences -> how many maximum MPNN sequences per trajectory to save if several pass filters
max_tm-score_filter -> filter out final lower ranking designs by this TM score cut off relative to all passing designs
max_seq-similarity_filter -> filter out final lower ranking designs by this sequence similarity cut off relative to all passing designs
sampling_temp = 0.1 -> sampling temperature for amino acids, T=0.0 means taking argmax, T>>1.0 means sampling randomly.")
# MPNN settings - advanced
sample_seq_parallel -> how many sequences to sample in parallel, reduce if running out of memory
backbone_noise -> backbone noise during sampling, 0.00-0.02 are good values
model_path -> path to the MPNN model weights
mpnn_weights -> whether to use "original" mpnn weights or "soluble" weights
save_mpnn_fasta -> whether to save MPNN sequences as fasta files, normally not needed as the sequence is also in the CSV file
# AF2 design settings - advanced
num_recycles_design -> how many recycles of AF2 for design
num_recycles_validation -> how many recycles of AF2 use for structure prediction and validation
optimise_beta -> optimise predictions if beta sheeted trajectory detected?
optimise_beta_extra_soft -> how many extra soft iterations to add if beta sheets detected
optimise_beta_extra_temp -> how many extra temporary iterations to add if beta sheets detected
optimise_beta_recycles_design -> how many recycles to do during design if beta sheets detected
optimise_beta_recycles_valid -> how many recycles to do during reprediction if beta sheets detected
# Optimise script
remove_unrelaxed_trajectory -> remove the PDB files of unrelaxed designed trajectories, relaxed PDBs are retained
remove_unrelaxed_complex -> remove the PDB files of unrelaxed predicted MPNN-optimised complexes, relaxed PDBs are retained
remove_binder_monomer -> remove the PDB files of predicted binder monomers after scoring to save space
zip_animations -> at the end, zip Animations trajectory folder to save space
zip_plots -> at the end, zip Plots trajectory folder to save space
save_trajectory_pickle -> save pickle file of the generated trajectory, careful, takes up a lot of storage space!
max_trajectories -> how many maximum trajectories to generate, for benchmarking
acceptance_rate -> what fraction of trajectories should yield designs passing the filters, if the proportion of successful designs is less than this fraction then the script will stop and you should adjust your design weights
start_monitoring -> after what number of trajectories should we start monitoring acceptance_rate, do not set too low, could terminate prematurely
# debug settings
enable_mpnn = True -> whether to enable MPNN design
enable_rejection_check -> enable rejection rate check
Voici les fonctionnalités par lesquelles vos conceptions seront filtrées. Si vous ne souhaitez pas en utiliser, définissez simplement null comme seuil. L'option plus élevée indique si les valeurs supérieures au seuil doivent être conservées (vrai) ou inférieures (faux). Les fonctionnalités commençant par N_ correspondent aux statistiques de chaque modèle AlphaFold. Les moyennes sont prédites pour tous les modèles.
MPNN_score -> MPNN sequence score, generally not recommended as it depends on protein
MPNN_seq_recovery -> MPNN sequence recovery of original trajectory
pLDDT -> pLDDT confidence score of AF2 complex prediction, normalised to 0-1
pTM -> pTM confidence score of AF2 complex prediction, normalised to 0-1
i_pTM -> interface pTM confidence score of AF2 complex prediction, normalised to 0-1
pAE -> predicted alignment error of AF2 complex prediction, normalised compared AF2 by n/31 to 0-1
i_pAE -> predicted interface alignment error of AF2 complex prediction, normalised compared AF2 by n/31 to 0-1
i_pLDDT -> interface pLDDT confidence score of AF2 complex prediction, normalised to 0-1
ss_pLDDT -> secondary structure pLDDT confidence score of AF2 complex prediction, normalised to 0-1
Unrelaxed_Clashes -> number of interface clashes before relaxation
Relaxed_Clashes -> number of interface clashes after relaxation
Binder_Energy_Score -> Rosetta energy score for binder alone
Surface_Hydrophobicity -> surface hydrophobicity fraction for binder
ShapeComplementarity -> interface shape complementarity
PackStat -> interface packstat rosetta score
dG -> interface rosetta dG energy
dSASA -> interface delta SASA (size)
dG/dSASA -> interface energy divided by interface size
Interface_SASA_% -> Fraction of binder surface covered by the interface
Interface_Hydrophobicity -> Interface hydrophobicity fraction of binder interface
n_InterfaceResidues -> number of interface residues
n_InterfaceHbonds -> number of hydrogen bonds at the interface
InterfaceHbondsPercentage -> number of hydrogen bonds compared to interface size
n_InterfaceUnsatHbonds -> number of unsatisfied buried hydrogen bonds at the interface
InterfaceUnsatHbondsPercentage -> number of unsatisfied buried hydrogen bonds compared to interface size
Interface_Helix% -> proportion of alfa helices at the interface
Interface_BetaSheet% -> proportion of beta sheets at the interface
Interface_Loop% -> proportion of loops at the interface
Binder_Helix% -> proportion of alfa helices in the binder structure
Binder_BetaSheet% -> proportion of beta sheets in the binder structure
Binder_Loop% -> proportion of loops in the binder structure
InterfaceAAs -> number of amino acids of each type at the interface
HotspotRMSD -> unaligned RMSD of binder compared to original trajectory, in other words how far is binder in the repredicted complex from the original binding site
Target_RMSD -> RMSD of target predicted in context of the designed binder compared to input PDB
Binder_pLDDT -> pLDDT confidence score of binder predicted alone
Binder_pTM -> pTM confidence score of binder predicted alone
Binder_pAE -> predicted alignment error of binder predicted alone
Binder_RMSD -> RMSD of binder predicted alone compared to original trajectory
Merci à Lennart Nickel, Yehlin Cho, Casper Goverde et Sergey Ovchinnikov pour leur aide dans le codage et la discussion d'idées. Ce référentiel utilise le code de :


