binnacle
1.0.0
À
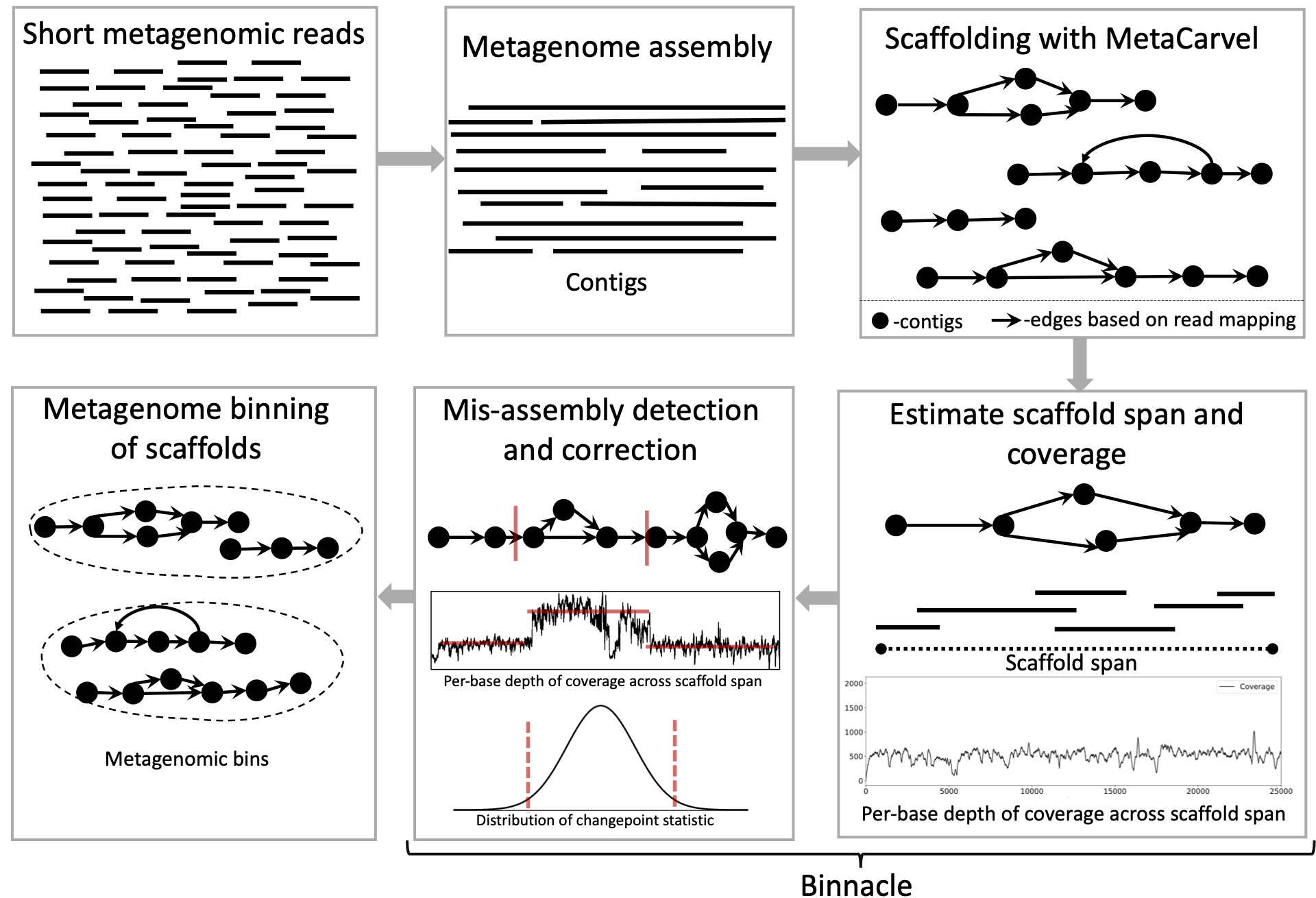
Binnacle calcule avec précision la couverture des échafaudages de graphiques et s'intègre de manière transparente aux principales méthodes de binning telles que Metabat2, Maxbin 2.0 et Concoct. L'utilisation d'échafaudages graphiques, par opposition aux contigs (approche la plus courante) pour le binning améliore la contiguïté et la qualité des bacs métagénomiques et peut capturer un ensemble plus large des éléments accessoires des génomes reconstruits.
Pour exécuter Binnacle, vous aurez besoin de Python 3.7.x, BedTools, Samtools, Biopython, Networkx, Numpy et Pandas.
Un fichier Environment.yml est disponible et cela peut être utilisé pour créer un environnement conda qui est adapté à l'exécution du binnacle. La documentation détaillée sur la façon d'installer ces packages est donnée ici. Nous utilisons des échafaudages graphiques qui sont sortis de l'outil d'échafaudage Metacarvel, vous devrez donc également télécharger et installer Metacarvel. Il existe un guide d'installation étape par étape pour Metacarvel.
Généralement, lorsque vous avez un ou plusieurs échantillons métagénomiques, nous devons assembler, échafaudages et contigus / échafaudages de bacs de chaque échantillon pour générer des bacs métagénomiques. Nous vous recommandons d'utiliser Megahit pour l'assemblage et Metacarvel pour l'échafaudage. Nous fournissons un guide d'assistance pour travailler à travers des étapes d'assemblage d'assemblage, d'échafaudage et de couverture par base ici.
Suivez ces étapes pour générer des fichiers pour exécuter des méthodes de binning avec des échafaudages graphiques:
python Estimate_Abundances.py -g [ORIENTED.gml] -a [COVERAGE_SORTED.txt] -c [CONTIGS.fa] -d [OUTPUT_DIRECTORY]
usage: Estimate_Abundances.py [-h] [-g ASSEMBLY] [-a COVERAGE] [-bam BAMFILE]
[-bed BEDFILE] [-c CONTIGS] -d DIR [-o COORDS]
[-w WINDOW_SIZE] [-t THRESHOLD]
[-n NEIGHBOR_CUTOFF] [-p POSCUTOFF]
[-pre PREFIX]
binnacle: A tool for binning metagenomic datasets using assembly graphs and
scaffolds generated by metacarvel. Estimate_Abundances.py estimates abundance
for scaffolds generated by MetaCarvel. If the coordinates computed by binnacle
is specified then the abundance for each scaffold is estimated based on the
contig abundances and the coordinates. If the coordinates are not specified
then binnacle etimates the abundance from scratch. While calculating all vs
all abundances please specify the coordinates(Coordinates_After_Delinking.txt)
through the "coords" parameter. The abundances can be provided as a bed file,
bam file or a text file describing the per base coverage obtained by running
the genomeCoverageBed program of the bedtools suite.
optional arguments:
-h, --help show this help message and exit
-g ASSEMBLY, --assembly ASSEMBLY
Assembly Graph generated by Metacarvel
-a COVERAGE, --coverage COVERAGE
Output generated by running genomecov -d on the bed
file generated by MetaCarvel.
-bam BAMFILE, --bamfile BAMFILE
Bam file from aligning reads to contigs
-bed BEDFILE, --bedfile BEDFILE
Bed file from aligning reads to contigs. If bed file
is provided please provide a fasta file of the contigs
-c CONTIGS, --contigs CONTIGS
Contigs generated by the assembler, contigs.fasta
-d DIR, --dir DIR output directory for results
-o COORDS, --coords COORDS
Coordinate file generated by Binnacle
-w WINDOW_SIZE, --window_size WINDOW_SIZE
Size of the sliding window for computing test
statistic to identify changepoints in coverages
(Default=1500)
-t THRESHOLD, --threshold THRESHOLD
Threshold to identify outliers (Default=99)
-n NEIGHBOR_CUTOFF, --neighbor_cutoff NEIGHBOR_CUTOFF
Filter size to identify outliers within (Defualt=100)
-p POSCUTOFF, --poscutoff POSCUTOFF
Position cutoff to consider delinking (Default=100)
-pre PREFIX, --prefix PREFIX
Prefix to be attached to all outputs
-g Path to oriented.gml from running metacarvel on sample
-c Path to contigs obtained by assembling reads of sample
-a Coverage of contigs in ths sample by mapping to its reads -- See Wiki for how to calculate coverage information
-d Output directory
-a Coverage of contigs in Sample 1 by mapping reads of Sample 2 -- See Wiki for how to calculate coverage information
-o Coordinates of scaffolds from Sample 1 that you would have generated from the previous step.
-d Same output directory as Sample 1
python Collate.py -h
usage: Collate.py [-h] -d DIR [-m METHOD] [-k KEEP]
binnacle: A tool for binning metagenomic datasets using assembly graphs and
scaffolds generated by metacarvel.Estimate_Abundances.py estimates abundance
for scaffolds generated by MetaCarvel. The program Collate.py collects the
summary files generated by Estimate_Abundances.py
optional arguments:
-h, --help show this help message and exit
-d DIR, --dir DIR Output directory that contains the summary files
generated by running Estimate_Abundances.py
-m METHOD, --method METHOD
Binning method to format the output to. Presently we
support 1. Metabat 2. Maxbin 3. Concoct 4. Binnacle
(Default)
-k KEEP, --keep KEEP Retain the summary files generated by
Estimate_Abundances.py. Defaults to True
Veuillez vérifier le wiki pour une description détaillée de la configuration de l'environnement Python, des méthodes pour calculer la couverture et un flux de travail typique pour exécuter le binnacle.
Pour visualiser les échafaudages de graphiques, nous recommandons d'utiliser le métagénomescope qui est un navigateur Web. L'entrée de MetageNomeScope est assembly_graph_filtered.gml. Une documentation détaillée sur l'installation et l'exécution du métagénomescope est donnée ici.
Veuillez citer Muralidharan HS, Shah N, Meisel JS et Pop M (2021) Binnacle: Utilisation d'échafaudages pour améliorer la contiguïté et la qualité des bacs métagénomiques. Devant. Microbiol. 12: 638561. doi: 10.3389 / fmicb.2021.638561.
Cet outil est toujours en cours de développement. Veuillez ouvrir le problème ici sur GitHub ou contactez-nous si vous avez des questions.
Harihara Muralidharan: [email protected]
Nidhi Shah: [email protected]


