ClockstaR
1.0.0

Sebastian Duchene, Martyna Molak, dan Simon YW Ho.
Laboratorium Ekologi Molekuler, Evolusi, dan Filogenetika (MEEP).
Sekolah Ilmu Biologi
Universitas Sydney
10 Juni 2015
partition_data_partitionfinder('drag fasta file with concatenated data here', 'drag partition finder output here')
optim.trees.interactive(folder.parts = 'path to your folder with fasta files and tree topology here')
Menerapkan optimasi jarak pohon menggunakan turunan jarak BSD
Menerapkan versi paralel untuk jarak topologi
Tulis tutorial untuk pengelompokan jarak topologi
Integrasikan generator model untuk pengujian model
Integrasikan RaxML untuk optimalisasi kemungkinan maksimum panjang cabang dan topologi
Memperkirakan rentang waktu evolusi dengan kumpulan data multigene adalah praktik umum dalam studi filogenetik. Kumpulan data multigen dapat dipartisi berdasarkan gen, posisi kodon, atau keduanya. Dalam tutorial ini, kami mengacu pada “subkumpulan data” sebagai gen individu atau subunit apa pun dari kumpulan data multigen. Istilah "partisi" mengacu pada sekelompok subset data.
Meskipun subset data dapat digabungkan dan dianalisis dengan model jam santai tunggal, pola variasi laju antar garis keturunan dapat berbeda antar subset data bahkan ketika topologi pohonnya identik. Misalnya, variasi laju antar garis keturunan pada gen mitokondria dapat berbeda dengan variasi laju gen inti. Oleh karena itu, model jam santai yang berbeda dapat ditugaskan ke subkumpulan data yang berbeda untuk meningkatkan perkiraan rentang waktu evolusi dan kesesuaian statistik (lihat Duchene dan Ho., 2014a)
Ada banyak cara untuk mempartisi kumpulan data multigene. Pendekatan umum untuk membandingkan skema partisi adalah dengan menggunakan faktor Bayes atau kriteria berbasis kemungkinan untuk kesesuaian model. Namun dalam sebagian besar kasus, pengujian semua kemungkinan skema partisi tidak dapat dilakukan, terutama dengan metode penghitungan faktor Bayes yang intensif komputasi.
ClockstaR memperkirakan panjang cabang filogenetik dari setiap subset data. Jarak skor cabang, yang dikenal sebagai sBSDmin, dihitung untuk setiap pasangan pohon sebagai ukuran perbedaan pola variasi laju antar-garis keturunan. Jarak ini digunakan untuk menyimpulkan strategi partisi terbaik menggunakan statistik GAP dengan algoritma clustering PAM, seperti yang diterapkan dalam cluster paket (Maechler et al., 2012) (untuk detail metrik sBSDmin, lihat Duchene et al., 2014b) .
ClockstaR adalah paket R untuk analisis jam molekuler filogenetik dari kumpulan data multi-gen. Ia menggunakan pola variasi tingkat garis keturunan untuk gen yang berbeda untuk memilih strategi partisi jam. Metode ini menggunakan metrik jarak pohon filogenetik dan algoritme pembelajaran mesin tanpa pengawasan untuk mengidentifikasi jumlah partisi jam yang optimal, dan gen mana yang harus dianalisis pada setiap partisi. Strategi partisi yang dipilih di ClocsktaR dapat digunakan untuk analisis jam molekuler selanjutnya dengan program seperti BEAST, MrBayes, PhyloBayes, dan lainnya.
Silakan ikuti tautan ini untuk publikasi aslinya.
ClockstaR memerlukan instalasi R. Ini juga memerlukan beberapa dependensi R, yang dapat diperoleh melalui R, seperti dijelaskan di bawah.
Silakan kirimkan permintaan atau pertanyaan apa pun ke Sebastian Duchene (sebastian.duchene[at]sydney.edu.au). Beberapa perangkat lunak dan sumber daya lain dapat ditemukan di Laboratorium Ekologi Molekuler, Evolusi, dan Filogenetika di Universitas Sydney.
Unduh repositori ini sebagai file zip dan unzip. Petunjuk berikut menggunakan folder clockstar_example_data, yang berisi beberapa file fasta dan pohon filogenetik dalam format newick. Buka salah satu file ini dalam editor teks, seperti pengatur teks. Data ini disimulasikan berdasarkan empat pola variasi laju evolusi. Perhatikan bahwa pohon adalah topologi pohon untuk semua gen, atau partisi data. Untuk menjalankan ClockstaR harap format data Anda seperti contoh data di clockstar_example_data.
ClockstaR dapat diinstal langsung dari GitHub. Ini memerlukan paket devtools. Ketik kode berikut pada prompt R untuk menginstal semua alat yang diperlukan (perhatikan Anda memerlukan koneksi internet untuk mengunduh paket secara langsung):.
install . packages ( " devtools " )
library (devtools)
install_github ( ' ClockstaR ' , ' sebastianduchene ' )Setelah mengunduh dan menginstal, muat ClockstaR dengan perpustakaan fungsi.
library (ClockstaR2)Untuk melihat contoh bagaimana program dijalankan ketik:
example (ClockstaR2)Sisa tutorial ini menggunakan folder clockstar_example_data
Langkah pertama adalah mendapatkan pohon gen untuk setiap penyelarasan. Untuk melakukan ini, kami menggunakan topologi pohon, dan mengoptimalkan panjang cabang menggunakan masing-masing penyelarasan gen individu, dalam hal ini A1.fasta hingga C3.fasta. Jika Anda memiliki pohon gen, simpan dalam format newick dalam file dan lanjutkan ke langkah berikutnya (menjalankan clockstar secara interaktif).
Ketik kode berikut di prompt R dan tekan enter:
optim . trees . interactive ()Jika Anda mendapatkan pesan kesalahan tentang menginstal paket phangorn, silakan gunakan kode ini lalu ulangi optim.trees.interactive()
install . packcages ( " phangorn " )ClockstaR akan mencetak pesan berikut:
Please drag a folder with the data subsets and a tree topology . The files should be in FASTA format, and the trees in NEWICKSeret folder clockstar_example_data ke konsol R dan ketik enter. Perhatikan bahwa folder tersebut hanya boleh berisi penyempurnaan dalam format FASTA, dan topologi pohon dalam format NEWICK. Anda akan melihat pesan berikut:
What should be the name of the file to save the optimised trees ?Ketikkan nama file untuk pohon yang dioptimalkan. Dalam hal ini kita akan menggunakan "example.trees"
example . treesPada titik ini, ClockstaR akan menanyakan apakah harus menggunakan model substitusi terpisah untuk setiap gen, atau menggunakan JC di semua kasus. Karena data ini disimulasikan di bawah JC, kita akan mengetik "n" dan tekan enter. Ketik "y" untuk menentukan setiap model substitusi secara terpisah.
Setelah mengetik "n" dan menekan enter, ClockstaR akan mulai berjalan. Ini akan mencetak pohon gen di perangkat grafis. Jika pohon yang ditentukan telah di-root, mungkin juga akan muncul beberapa peringatan, yang dapat diabaikan dengan aman.
Buka folder clockstar_example_data. Anda akan menemukan file dengan nama "example.trees", seperti yang ditentukan beberapa langkah di atas. Buka example.trees di editor teks. Ini berisi setiap pohon gen dan nama pohon, sesuai dengan nama keselarasan gen. Seharusnya terlihat seperti ini:
A1 . fasta (( t1 : 0.01504695462 ,( t2 : 0.00987 ...
A2 . fasta (( t1 : 0.01520523401 ,( t2 : 0.01317 ...
A3 . fasta (( t1 : 0.01519309467 ,( t2 : 0.01092 ...
.
.
.File dengan pohon ini akan digunakan untuk langkah berikutnya.
Untuk langkah ini perlu adanya pohon gen dalam sebuah file, seperti yang diperoleh pada langkah sebelumnya.
Buka R dan muat ClockstaR seperti yang ditunjukkan di atas. Ketik kode berikut saat diminta:
clockstar . interactive ()ClockstaR akan mencetak pesan berikut:
please drag or type in the path to your gene trees file in NEWICK format :Seret file dengan pohon gen ke konsol R. Jika Anda mengikuti langkah sebelumnya, file tersebut akan diberi nama example.trees. Ketik masuk.
Tergantung pada paket yang telah Anda instal, ClockstaR mungkin menanyakan apakah paket tersebut harus dijalankan secara paralel. Ini efisien untuk kumpulan data besar. Namun untuk contoh datanya tidak akan terlalu berpengaruh, jadi ketikkan "n" jika Anda melihat pesan ini lalu ketikkan enter:
Packages foreach and doParallel are available for parallel computation
Should we run ClockstaR in parallel (y / n) ? (This is good for large data sets)Clockstar sekarang akan mulai berjalan. Output di layar akan terlihat seperti ini:
[ 1 ] " Calculating sBSDmin distances between all pairs of trees "
[ 1 ] " Estimating tree distances "
[ 1 ] " estimating distances 1 of 11 "
[ 1 ] " estimating distances 2 of 11 "
[ 1 ] " estimating distances 3 of 11 "
[ 1 ] " estimating distances 4 of 11 "
[ 1 ] " estimating distances 5 of 11 "
.
.
.Setelah memperkirakan jarak pohon (dijelaskan dalam publikasi asli), ClockstaR akan mencetak pesan berikut:
" I finished calculating the sBSDmin distances between trees "
The settings for clustering with ClockstaR are :
PAM clustering algorithm
K from 1 to number of data subsets - 1
SEmax criterion to select the optimal k
500 bootstrap replicates
Are these correct ? (y / n)Ini adalah pengaturan untuk algoritma clustering. Mereka sesuai untuk sebagian besar kumpulan data, jadi dalam contoh ini kita dapat mengetik "y" lalu enter. Dengan mengetik "n" kita dapat mengubah pengaturan ini, untuk lebih jelasnya lihat Kaufman dan Rousseeuw (2009).
ClockstaR sekarang akan menjalankan algoritma clustering. Pada akhirnya akan mencetak jumlah partisi terbaik dan menanyakan apakah hasilnya harus disimpan dalam file pdf:
[ 1 ] " ClockstaR has finished running "
[ 1 ] " The best number of partitions for your data set is: 3 "
Do you wish to save the results in a pdf file ? (y / n)Ketik "y" lalu enter.
ClockstaR kemudian akan menanyakan nama file keluaran:
What should be the name and path of the output file ?Untuk contoh ini ketik "example_run" dan enter, tetapi nama apa pun dapat digunakan.
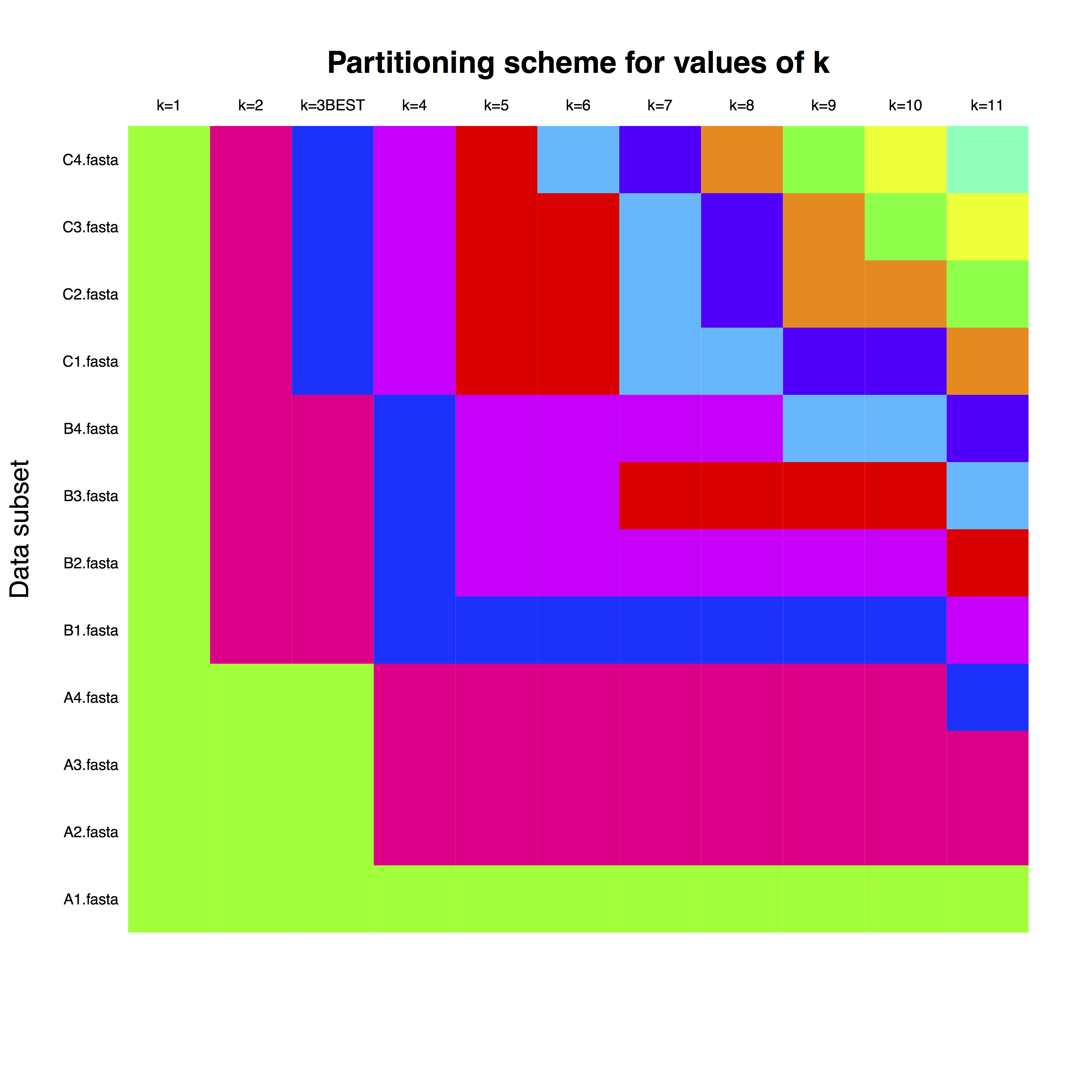
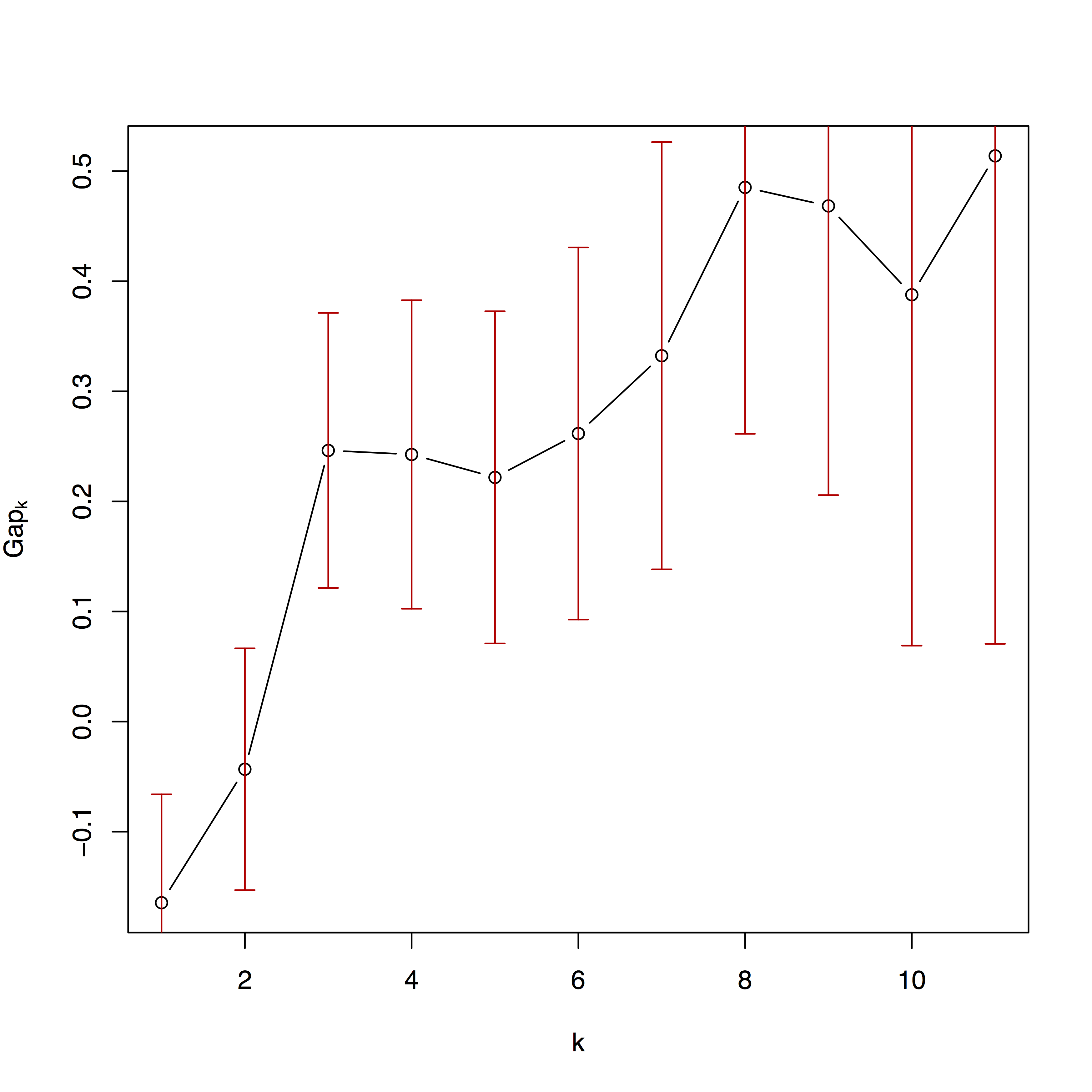
Sekarang buka folder clockstar_example_data dan buka dua file pdf, example_run_gapstats.pdf dan example_run_matrix.pdf.
example_run_matrix adalah matriks, di mana baris-barisnya berhubungan dengan setiap gen, seperti yang disebutkan dalam file FASTA. Kolom adalah jumlah partisi, dan warna mewakili penempatan setiap gen pada partisi jam. Misalnya, untuk k =3, yang merupakan jumlah partisi terbaik, seseorang dapat menggunakan partisi jam terpisah untuk gen dengan huruf A, B, dan C.
Plot kedua adalah kesesuaian algoritma pengelompokan di sejumlah partisi yang berbeda. Rincian lebih lanjut tersedia di Kaufman dan Rousseeuw (2009) dan dalam dokumentasi untuk cluster paket.
ClockstaR dapat dijalankan dengan pengaturan khusus lainnya. Silakan lihat dokumentasi untuk detail lainnya atau hubungi saya untuk pertanyaan apa pun di sebastian.duchene[at]sydney.edy.au.
Logonya dirancang oleh Jun Tong
Duchene, S., & Ho, SY (2014a). Menggunakan beberapa model jam santai untuk memperkirakan rentang waktu evolusi dari data urutan DNA. Filogenetika dan Evolusi Molekuler (77): 65-70.
Duchene, S., Molak, M., & Ho, SY (2014b). ClockstaR: memilih jumlah model jam santai dalam analisis filogenetik molekuler. Bioinformatika 30 (7): 1017-1019.
Kaufman, L., & Rousseeuw, PJ (2009). Menemukan kelompok dalam data: pengantar analisis klaster (Vol. 344). John Wiley & Putra.


