BRAKER
v3.0.8
Berikut ini adalah rekaman sesi lokakarya BGA23 pertama di Braker. Jika belajar dengan menonton video itu mudah bagi Anda, pertimbangkan untuk menonton itu: https://www.youtube.com/watch?v=uxtkj4mukyg
Braker3 sekarang di https://usegalaxy.eu/
Tsebra & Braker3 Terkait:
Braker & Augustus terkait:
Genemark terkait:
Mark Borodovsky, Georgia Tech, AS, [email protected]
Tomas Bruna, Genome Genome Institute, AS, [email protected]
Alexandre Lomsazde, Georgia Tech, AS, [email protected]
[A] University of Greifswald, Institute for Mathematics and Computer Science, Walther-Rathenau-Str. 47, 17489 Greifswald, Jerman
[b] University of Greifswald, Pusat Genomik Fungsional Mikroba, Felix-Hausdorff-Str. 8, 17489 Greifswald, Jerman
[C] Gabungan Georgia Tech dan Emory University Wallace H Coulter Departemen Teknik Biomedis, 30332 Atlanta, USA
[D] Sekolah Sains dan Teknik Komputasi, 30332 Atlanta, USA
[E] Institut Fisika dan Teknologi Moskow, Wilayah Moskow 141701, Dolgoprudny, Rusia
![Braker2-Team-2 [Gbr10]](https://images.downcodes.com/uploads/20250214/img_67aee79a0cb7530.png)
![Braker2-Team-1 [Fig11]](https://images.downcodes.com/uploads/20250214/img_67aee79a0d1eb31.png)
![Braker2-Team-3 [Gbr12]](https://images.downcodes.com/uploads/20250214/img_67aee79a0da9c32.png)
![Braker2-Team-4 [Fig13]](https://images.downcodes.com/uploads/20250214/img_67aee79a0e49933.png)
Gambar 1: Penulis Braker saat ini, dari kiri ke kanan: Mario Stanke, Alexandre Lomsadze, Katharina J. Hoff, Tomas Bruna, Lars Gabriel, dan Mark Borodovsky. Kami mengakui bahwa komunitas ilmuwan yang lebih besar berkontribusi pada Kode Braker (misalnya melalui permintaan pull).
Pengembangan Braker1, Braker2, dan Braker3 didukung oleh National Institutes of Health (NIH) [GM128145 ke MB dan MS]. Pengembangan Braker3 sebagian didanai oleh kompetensi data proyek yang diberikan kepada KJH dan MS oleh Pemerintah Mecklenburg-Vorpommern, Jerman.
Pemilih transkrip untuk Braker (TSebra) tersedia di https://github.com/gaius-augustus/tsebra.
GeneMark-ETP, salah satu pencari gen di inti Braker, tersedia di https://github.com/gatech-genemark/genemark-etp.
Augustus, pencari gen kedua di inti Braker, tersedia di https://github.com/gaius-augustus/augustus.
Galba, spin-off pipa Braker untuk menggunakan miniprot atau genomethreader untuk menghasilkan gen pelatihan, tersedia di https://github.com/gaius-ugustus/galba.
Jumlah genom berurutan yang berkembang pesat membutuhkan metode yang sepenuhnya otomatis untuk anotasi struktur gen yang akurat. Dengan tujuan ini, kami telah mengembangkan Braker1 R1 R0 , kombinasi Genemark-ET R2 dan Augustus R3, R4 , yang menggunakan data genomik dan RNA-seq untuk secara otomatis menghasilkan anotasi struktur gen penuh dalam genom baru.
Namun, kualitas data RNA-seq yang tersedia untuk anotasi genom baru adalah variabel, dan dalam beberapa kasus, data RNA-seq tidak tersedia sama sekali.
Braker2 adalah perpanjangan dari Braker1 yang memungkinkan pelatihan sepenuhnya otomatis dari alat prediksi gen Genemark-es/ET/EP/ETP R14, R15, R17, F1 dan Augustus dari RNA-seq dan/atau informasi homologi protein, dan yang mengintegrasikan Bukti ekstrinsik dari RNA-seq dan informasi homologi protein ke dalam prediksi .
Berbeda dengan metode lain yang tersedia yang mengandalkan informasi homologi protein, Braker2 mencapai akurasi prediksi gen yang tinggi bahkan dengan tidak adanya anotasi spesies yang sangat terkait dan tanpa adanya data RNA-seq.
Braker3 adalah pipa terbaru di Braker Suite. Ini memungkinkan penggunaan data RNA-seq dan protein dalam pipa yang sepenuhnya otomatis untuk melatih dan memprediksi gen yang sangat andal dengan Genemark-ETP dan Augustus. Hasil pipa adalah set gen gabungan dari kedua alat prediksi gen, yang hanya mengandung gen dengan dukungan yang sangat tinggi dari bukti ekstrinsik.
Dalam panduan pengguna ini, kami akan merujuk ke Braker1, Braker2, dan Braker3 hanya sebagai Braker karena mereka dieksekusi oleh skrip yang sama ( braker.pl ).
Gunakan perakitan genom berkualitas tinggi. Jika Anda memiliki sejumlah besar perancah yang sangat singkat dalam perakitan genom Anda, perancah pendek itu kemungkinan akan meningkatkan runtime secara dramatis tetapi tidak akan meningkatkan akurasi prediksi.
Gunakan nama perancah sederhana dalam file genome (misalnya >contig1 akan bekerja lebih baik dari >contig1my custom species namesome putative function /more/information/ and lots of special characters %&!*(){} ). Buat nama perancah di semua file fasta Anda sederhana sebelum menjalankan program penyelarasan apa pun.
Untuk memprediksi gen secara akurat dalam genom baru, genom harus ditutupi untuk pengulangan. Ini akan menghindari prediksi struktur gen positif palsu di daerah yang berulang dan rendah. Mengulangi masking juga penting untuk memetakan data RNA-seq ke genom dengan beberapa alat (pemetaan RNA-seq lainnya, seperti HISAT2, mengabaikan informasi penyemburan). Dalam kasus Genemark-ES/ET/EP/ETP dan Augustus, softmasking (yaitu menempatkan daerah berulang ke dalam huruf kecil dan semua daerah lain ke dalam huruf besar) mengarah ke hasil yang lebih baik daripada hardmasking (yaitu mengganti huruf di daerah berulang dengan surat tersebut N untuk nukleotida yang tidak diketahui).
Banyak genom memiliki struktur gen yang akan diprediksi secara akurat dengan parameter standar Genemark-ES/ET/EP/ETP dan Augustus di dalam Braker. Namun, beberapa genom memiliki fitur spesifik clade, yaitu model titik cabang khusus dalam jamur, atau pola lokasi sambungan non-standar. Harap baca bagian Opsi [Opsi] untuk menentukan apakah ada opsi khusus yang dapat meningkatkan akurasi prediksi gen dalam genom spesies target Anda.
Selalu periksa hasil prediksi gen sebelum penggunaan lebih lanjut! Anda dapat EG menggunakan browser genom untuk inspeksi visual model gen dalam konteks dengan data bukti ekstrinsik. Braker mendukung pembuatan hub data track untuk browser genom UCSC dengan Makehub untuk tujuan ini.
Braker terutama menampilkan data bukti ekstrinsik semi-unsupervised (RNA-seq dan/atau protein yang disambungkan) mendukung pelatihan Genemark-ES/ET/EP/ETP [F1] dan pelatihan Augustus selanjutnya dengan integrasi bukti ekstrinsik di final Langkah Prediksi Gen. Namun, sekarang ada sejumlah pipa tambahan yang termasuk dalam Braker. Berikut ini, kami memberikan gambaran tentang kemungkinan file input dan pipa:
![braker2-main-a [gbr1]](https://images.downcodes.com/uploads/20250214/img_67aee79a0eaf534.png)
Gambar 2: Braker Pipeline A: Pelatihan Genemark-es pada data genom, hanya; prediksi gen ab initio withaugustus
![braker2-main-b [gbr2]](https://images.downcodes.com/uploads/20250214/img_67aee79a0f13f35.png)
Gambar 3: Braker Pipeline B: Pelatihan GeneMark-ET didukung oleh RNA-seq informasi penyelarasan yang disambung, prediksi dengan Augustus dengan informasi penyelarasan yang sama.
![Braker2-Main-C [Gbr3]](https://images.downcodes.com/uploads/20250214/img_67aee79a0fa1036.png)
Gambar 4: Braker Pipeline C: Pelatihan Genemark-EP+ pada perataan yang disambung protein, informasi mulai dan hentikan, prediksi dengan Augustus dengan informasi yang sama, selain petunjuk CDSpart yang dirantai. Protein yang digunakan di sini dapat berjarak evolusioner ke organisme target.
![braker3-main-a [gbr4]](https://images.downcodes.com/uploads/20250214/img_67aee79a1010b37.png)
Gambar 5: Braker Pipeline D: Jika perlu, unduh, dan penyelarasan set RNA-seq untuk spesies target. Pelatihan GeneMark-ETP yang didukung oleh penyelarasan RNA-seq dan basis data protein besar (protein dapat berjarak evolusi). Selanjutnya, pelatihan dan prediksi Augustus menggunakan informasi ekstrinsik yang sama bersama-sama dengan hasil Genemark-ETP. Prediksi terakhir adalah kombinasi Tsebra dari hasil Augustus dan Genemark-ETP.
Kami sadar bahwa pemasangan "manual" dari Braker3 dan semua ketergantungannya membosankan dan sangat menantang tanpa izin akar. Oleh karena itu, kami menyediakan wadah Docker yang telah dikembangkan untuk dijalankan dengan singularitas. Semua informasi tentang wadah ini dapat ditemukan di https://hub.docker.com/r/teambraker/braker3
Singkatnya, bangun sebagai berikut:
singularity build braker3.sif docker://teambraker/braker3:latest
Mengeksekusi dengan:
singularity exec braker3.sif braker.pl
Tes dengan:
singularity exec -B $PWD:$PWD braker3.sif cp /opt/BRAKER/example/singularity-tests/test1.sh .
singularity exec -B $PWD:$PWD braker3.sif cp /opt/BRAKER/example/singularity-tests/test2.sh .
singularity exec -B $PWD:$PWD braker3.sif cp /opt/BRAKER/example/singularity-tests/test3.sh .
export BRAKER_SIF=/your/path/to/braker3.sif # may need to modify
bash test1.sh
bash test2.sh
bash test3.sh
Beberapa pengguna ingin menjalankan analisis mereka di dalam Docker (karena izin root diperlukan). Namun, jika itu tujuan Anda, Anda dapat menjalankan dan menguji wadah sebagai berikut
sudo docker run --user 1000:100 --rm -it teambraker/braker3:latest bash
bash /opt/BRAKER/example/docker-tests/test1.sh # BRAKER1
bash /opt/BRAKER/example/docker-tests/test2.sh # BRAKER2
bash /opt/BRAKER/example/docker-tests/test3.sh # BRAKER3
Semoga beruntung ;-)
$PATH Anda mungkin menyebabkan gangguan yang tidak terduga, menyebabkan kegagalan program. Harap pindahkan semua versi GeneMark yang lebih tua dari $PATH Anda (juga misalnya Genemark di ProtHint/dependencies ).
Pada saat rilis, versi Braker ini diuji dengan:
Augustus 3.5.0 F2
Genemark-ETP (Sumber Lihat Dockerfile)
BAMTOOLS 2.5.1 R5
Samtools 1.7-4-G93586ed R6
Spaln 2.3.3d R8, R9, R10
NCBI Blast+ 2.2.31+ R12, R13
Berlian 0.9.24
CDBFASTA 0.99
Cdbyank 0.981
Gushr 1.0.0
SRA Toolkit 3.00 R14
HISAT2 2.2.1 R15
Bedtools 2.30 R16
Stringtie2 2.2.1 R17
GFFREAD 0.12.7 R18
Compleasm 0.2.5 R27
Menjalankan Braker membutuhkan sistem Linux dengan bash dan Perl. Selain itu, Braker membutuhkan modul CPAN-Perl berikut untuk diinstal:
File::Spec::Functions
Hash::Merge
List::Util
MCE::Mutex
Module::Load::Conditional
Parallel::ForkManager
POSIX
Scalar::Util::Numeric
YAML
Math::Utils
File::HomeDir
Untuk Genemark-ETP, digunakan saat protein dan RNA-seq disediakan:
YAML::XSData::DumperThread::Queuethreads Di Ubuntu, misalnya, instal modul dengan CPANMINUS F4 : sudo cpanm Module::Name , misalnya sudo cpanm Hash::Merge .
Braker juga menggunakan modul Perl helpMod_braker.pm yang tidak tersedia di CPAN. Modul ini adalah bagian dari rilis Braker dan tidak memerlukan instalasi terpisah.
Jika Anda tidak memiliki izin root pada mesin Linux, coba siapkan lingkungan Anaconda (https://www.anaconda.com/distribution/) sebagai berikut:
wget https://repo.anaconda.com/archive/Anaconda3-2018.12-Linux-x86_64.sh
bash bin/Anaconda3-2018.12-Linux-x86_64.sh # do not install VS (needs root privileges)
conda install -c anaconda perl
conda install -c anaconda biopython
conda install -c bioconda perl-app-cpanminus
conda install -c bioconda perl-file-spec
conda install -c bioconda perl-hash-merge
conda install -c bioconda perl-list-util
conda install -c bioconda perl-module-load-conditional
conda install -c bioconda perl-posix
conda install -c bioconda perl-file-homedir
conda install -c bioconda perl-parallel-forkmanager
conda install -c bioconda perl-scalar-util-numeric
conda install -c bioconda perl-yaml
conda install -c bioconda perl-class-data-inheritable
conda install -c bioconda perl-exception-class
conda install -c bioconda perl-test-pod
conda install -c bioconda perl-file-which # skip if you are not comparing to reference annotation
conda install -c bioconda perl-mce
conda install -c bioconda perl-threaded
conda install -c bioconda perl-list-util
conda install -c bioconda perl-math-utils
conda install -c bioconda cdbtools
conda install -c eumetsat perl-yaml-xs
conda install -c bioconda perl-data-dumper
Selanjutnya instal Braker dan perangkat lunak lain "seperti biasa" saat berada di lingkungan Conda Anda. Catatan: Ada paket Bioconda Braker, dan paket Bioconda Augustus. Mereka bekerja. Tetapi mereka biasanya tertinggal di balik kode pengembangan kedua alat di GitHub. Karena itu kami merekomendasikan pemasangan manual dan penggunaan sumber terakhir.
Braker adalah kumpulan skrip Perl dan Python dan modul Perl. Skrip utama yang akan dipanggil untuk menjalankan Braker adalah braker.pl . Komponen Perl dan Python tambahan adalah:
align2hints.pl
filterGenemark.pl
filterIntronsFindStrand.pl
startAlign.pl
helpMod_braker.pm
findGenesInIntrons.pl
downsample_traingenes.pl
ensure_n_training_genes.py
get_gc_content.py
get_etp_hints.py
Semua skrip (file yang diakhiri dengan *.pl dan *.py ) yang merupakan bagian dari Braker harus dapat dieksekusi untuk menjalankan Braker. Ini seharusnya sudah terjadi jika Anda mengunduh Braker dari GitHub. Eksekusi dapat ditimpa jika Anda membatalkan transfer Braker pada USB-stick ke komputer lain. Untuk memeriksa apakah file yang diperlukan dapat dieksekusi, jalankan perintah berikut di direktori yang berisi skrip Braker Perl:
ls -l *.pl *.py
Outputnya harus mirip dengan ini:
-rwxr-xr-x 1 katharina katharina 18191 Mai 7 10:25 align2hints.pl
-rwxr-xr-x 1 katharina katharina 6090 Feb 19 09:35 braker_cleanup.pl
-rwxr-xr-x 1 katharina katharina 408782 Aug 17 18:24 braker.pl
-rwxr-xr-x 1 katharina katharina 5024 Mai 7 10:25 downsample_traingenes.pl
-rwxr-xr-x 1 katharina katharina 5024 Mai 7 10:23 ensure_n_training_genes.py
-rwxr-xr-x 1 katharina katharina 4542 Apr 3 2019 filter_augustus_gff.pl
-rwxr-xr-x 1 katharina katharina 30453 Mai 7 10:25 filterGenemark.pl
-rwxr-xr-x 1 katharina katharina 5754 Mai 7 10:25 filterIntronsFindStrand.pl
-rwxr-xr-x 1 katharina katharina 7765 Mai 7 10:25 findGenesInIntrons.pl
-rwxr-xr-x 1 katharina katharina 1664 Feb 12 2019 gatech_pmp2hints.pl
-rwxr-xr-x 1 katharina katharina 2250 Jan 9 13:55 log_reg_prothints.pl
-rwxr-xr-x 1 katharina katharina 4679 Jan 9 13:55 merge_transcript_sets.pl
-rwxr-xr-x 1 katharina katharina 41674 Mai 7 10:25 startAlign.pl
Penting bahwa x in -rwxr-xr-x hadir untuk setiap skrip. Jika bukan itu masalahnya, jalankan
`chmod a+x *.pl *.py`
Untuk mengubah atribut file.
Anda mungkin bermanfaat untuk menambahkan direktori di mana skrip Braker Perl berada di variabel lingkungan $PATH Anda. Untuk satu sesi bash, masukkan:
PATH=/your_path_to_braker/:$PATH
export PATH
Untuk membuat modifikasi $PATH ini tersedia untuk semua sesi bash, tambahkan baris di atas ke skrip startup (misalnya ~/.bashrc ).
Braker memanggil berbagai alat perangkat lunak bioinformatika yang bukan bagian dari Braker. Beberapa alat wajib, yaitu Braker tidak akan berjalan sama sekali jika alat ini tidak ada di sistem Anda. Alat lain adalah opsional. Harap instal semua alat yang diperlukan untuk menjalankan Braker dalam mode pilihan Anda.
Unduh GeneMark-ETP F1 dari http://github.com/gatech-genemark/genemark-etp atau https://topaz.gatech.edu/genemark/etp.for_braker.tar.gz. Buka dan instal GeneMark-ETP seperti yang dijelaskan dalam file README Genemark-ETP.
Jika sudah terkandung dalam variabel $PATH Anda, Braker akan menebak lokasi gmes_petap.pl atau gmetp.pl secara otomatis. Kalau tidak, Braker dapat menemukan GeneMark-ES/ET/EP/ETP Executable baik dengan menempatkannya di variabel lingkungan GENEMARK_PATH , atau dengan mengambil argumen baris perintah ( --GENEMARK_PATH=/your_path_to_GeneMark_executables/ ).
Untuk mengatur variabel lingkungan untuk sesi bash Anda saat ini, ketik:
export GENEMARK_PATH=/your_path_to_GeneMark_executables/
Tambahkan baris di atas ke skrip startup (misalnya ~/.bashrc ) untuk membuatnya tersedia untuk semua sesi bash.
Skrip Perl dalam GeneMark-ES/ET/EP/ETP dikonfigurasi dengan lokasi Perl default AT /usr/bin/perl .
Jika Anda menjalankan GeneMark-ES/ET/EP/ETP di lingkungan Anaconda (atau ingin menggunakan Perl dari variabel $PATH untuk alasan lain), ubah shebang dari semua skrip Genemark-ES/ET/EP/ETP dengan Perintah berikut yang terletak di dalam folder Genemark-ES/ET/EP/ETP:
perl change_path_in_perl_scripts.pl "/usr/bin/env perl"
Anda dapat memeriksa apakah GeneMark-ES/ET/EP diinstal dengan benar dengan menjalankan check_install.bash dan/atau mengeksekusi contoh dalam direktori GeneMark-E-tests .
Genemark-ETP kompatibel ke bawah, yaitu mencakup fungsionalitas Genemark-EP dan Genemark-ET di Braker juga.
Unduh Augustus dari cabang masternya di https://github.com/gaius-augustus/augustus. Buka Augustus dan instal Augustus menurut Augustus README.TXT . Jangan gunakan versi Augustus yang sudah ketinggalan zaman dari sumber lain, misalnya paket Debian atau paket Bioconda! Braker sangat tergantung pada khususnya pada direktori Augustus/skrip terkini, dan sumber-sumber lain sering tertinggal.
Anda harus menyusun Augustus pada sistem Anda sendiri untuk menghindari masalah dengan versi perpustakaan yang digunakan oleh Augustus. Instruksi kompilasi disediakan dalam file Augustus README.TXT ( Augustus/README.txt ).
Augustus terdiri dari augustus , alat prediksi gen, alat C ++ tambahan yang berlokasi di Augustus/auxprogs dan skrip Perl yang terletak di Augustus/scripts . Skrip Perl harus dapat dieksekusi (lihat instruksi di bagian komponen Braker.
Alat C ++ bam2hints adalah komponen penting dari Braker ketika dijalankan dengan RNA-seq. Sumber terletak di Augustus/auxprogs/bam2hints . Pastikan Anda menyusun bam2hints pada sistem Anda (harus secara otomatis dikompilasi ketika Augustus disusun, tetapi dalam hal masalah dengan bam2hints , silakan baca instruksi pemecahan masalah di Augustus/auxprogs/bam2hints/README ).
Karena Braker adalah pipa yang melatih Augustus, yaitu menulis file parameter spesifik spesies, Braker perlu menulis akses ke direktori konfigurasi Augustus yang berisi file tersebut ( Augustus/config/ ). Jika Anda menginstal Augustus secara global di sistem Anda, folder config biasanya tidak dapat ditulis oleh semua pengguna. Entah membuat direktori di mana config berada secara rekursif dapat ditulis oleh pengguna Augustus, atau menyalin config/ folder (rekursif) ke lokasi di mana pengguna memiliki izin menulis.
Augustus akan menemukan folder config dengan mencari variabel lingkungan $AUGUSTUS_CONFIG_PATH . Jika variabel lingkungan $AUGUSTUS_CONFIG_PATH tidak diatur, maka Braker akan melihat di jalur ../config relatif terhadap direktori di mana ia menemukan Augustus dieksekusi. Atau, Anda dapat memasok variabel sebagai argumen baris perintah untuk Braker ( --AUGUSTUS_CONFIG_PATH=/your_path_to_AUGUSTUS/Augustus/config/ ). Kami menyarankan Anda mengekspor variabel EG untuk sesi bash Anda saat ini:
export AUGUSTUS_CONFIG_PATH=/your_path_to_AUGUSTUS/Augustus/config/
Untuk membuat variabel tersedia untuk semua sesi bash, tambahkan baris di atas ke skrip startup, misalnya ~/.bashrc .
Silakan lihat DockerFile jika Anda ingin menginstal Augustus sebagai paket Debian. Sejumlah skrip perlu ditambal.
Braker mengharapkan seluruh direktori config Augustus di $AUGUSTUS_CONFIG_PATH , yaitu species subfolder dengan isinya (setidaknya generic ) dan extrinsic ! Memberikan folder yang dapat ditulis tetapi kosong di $AUGUSTUS_CONFIG_PATH tidak akan bekerja untuk Braker. Jika Anda perlu memisahkan Binary Augustus dan $AUGUSTUS_CONFIG_PATH , kami sarankan Anda secara rekursif menyalin konten konfigurasi yang tidak dapat ditulis ke lokasi yang dapat ditulis.
Jika Anda memiliki instalasi sistem Augustus di /usr/bin/augustus di seluruh sistem, salinan config yang tidak dapat dipenuhi AT /usr/bin/augustus_config/ . Folder /home/yours/ dapat Anda dapatkan. Salin dengan perintah berikut (dan juga atur variabel yang diperlukan kemudian):
cp -r /usr/bin/Augustus/config/ /home/yours/
export AUGUSTUS_CONFIG_PATH=/home/yours/augustus_config
export AUGUSTUS_BIN_PATH=/usr/bin
export AUGUSTUS_SCRIPTS_PATH=/usr/bin/augustus_scripts
Menambahkan direktori biner dan skrip Augustus ke variabel $PATH Anda memungkinkan sistem Anda untuk menemukan alat -alat ini, secara otomatis. Ini bukan persyaratan untuk menjalankan Braker untuk melakukan ini, karena Braker akan mencoba menebaknya dari lokasi variabel lingkungan lain ( $AUGUSTUS_CONFIG_PATH ), atau kedua direktori dapat disuplai sebagai argumen baris perintah ke braker.pl , tetapi kami merekomendasikan untuk Tambahkan ke variabel $PATH Anda. Untuk sesi bash Anda saat ini, ketik:
PATH=:/your_path_to_augustus/bin/:/your_path_to_augustus/scripts/:$PATH
export PATH
Untuk semua sesi bash Anda, tambahkan baris di atas ke skrip startup (misalnya ~/.bashrc ).
Di Ubuntu, Python3 biasanya diinstal secara default, python3 akan berada di variabel $PATH Anda, secara default, dan Braker akan secara otomatis menemukannya. Namun, Anda memiliki opsi untuk menentukan lokasi biner python3 dengan dua cara lain:
Ekspor variabel lingkungan $PYTHON3_PATH , misalnya dalam file ~/.bashrc Anda:
export PYTHON3_PATH=/path/to/python3/
Tentukan opsi baris perintah --PYTHON3_PATH=/path/to/python3/ to braker.pl .
Unduh Bamtools (misalnya git clone https://github.com/pezmaster31/bamtools.git ). Instal bamtools dengan mengetik berikut di shell Anda:
cd your-bamtools-directory mkdir build cd build cmake .. make
Jika sudah ada di variabel $PATH Anda, Braker akan menemukan bamtools, secara otomatis. Jika tidak, Braker dapat menemukan biner Bamtools baik dengan menggunakan variabel lingkungan $BAMTOOLS_PATH , atau dengan mengambil argumen baris perintah ( --BAMTOOLS_PATH=/your_path_to_bamtools/bin/ f6 ). Untuk mengatur variabel lingkungan misalnya untuk sesi bash Anda saat ini, ketik:
export BAMTOOLS_PATH=/your_path_to_bamtools/bin/
Tambahkan baris di atas ke skrip startup (misalnya ~/.bashrc ) untuk mengatur variabel lingkungan untuk semua sesi bash.
Anda dapat menggunakan NCBI Blast+ atau Diamond untuk menghilangkan gen pelatihan yang berlebihan. Anda tidak membutuhkan kedua alat. Jika ada berlian, itu akan lebih disukai karena jauh lebih cepat.
Dapatkan dan buka kemasan berlian sebagai berikut:
wget http://github.com/bbuchfink/diamond/releases/download/v0.9.24/diamond-linux64.tar.gz
tar xzf diamond-linux64.tar.gz
Jika sudah ada di variabel $PATH Anda, Braker akan menemukan Diamond, secara otomatis. Kalau tidak, Braker dapat menemukan biner berlian baik dengan menggunakan variabel lingkungan $DIAMOND_PATH , atau dengan mengambil argumen baris perintah ( --DIAMOND_PATH=/your_path_to_diamond ). Untuk mengatur variabel lingkungan misalnya untuk sesi bash Anda saat ini, ketik:
export DIAMOND_PATH=/your_path_to_diamond/
Tambahkan baris di atas ke skrip startup (misalnya ~/.bashrc ) untuk mengatur variabel lingkungan untuk semua sesi bash.
Jika Anda memutuskan untuk Blast+, instal NCBI Blast+ dengan sudo apt-get install ncbi-blast+ .
Jika sudah ada di variabel $PATH Anda, Braker akan menemukan BlastP, secara otomatis. Jika tidak, Braker dapat menemukan biner blastp baik dengan menggunakan variabel lingkungan $BLAST_PATH , atau dengan mengambil argumen baris perintah ( --BLAST_PATH=/your_path_to_blast/ ). Untuk mengatur variabel lingkungan misalnya untuk sesi bash Anda saat ini, ketik:
export BLAST_PATH=/your_path_to_blast/
Tambahkan baris di atas ke skrip startup (misalnya ~/.bashrc ) untuk mengatur variabel lingkungan untuk semua sesi bash.
Alat berikut diperlukan oleh GeneMark-ETP dan akan mencoba menemukannya di variabel $PATH Anda. Jadi pastikan untuk menambahkan lokasi mereka ke $PATH Anda, misalnya:
export PATH=$PATH:/your/path/to/Tool
Untuk semua alat di bawah ini, tambahkan baris di atas ke skrip startup (misalnya ~/.bashrc ) untuk memperpanjang variabel $PATH Anda untuk semua sesi bash.
Alat perangkat lunak ini hanya wajib jika Anda menjalankan Braker dengan data RNA-seq dan protein!
Stringtie2 digunakan oleh GeneMark-ETP untuk mengumpulkan keberpihakan RNA-seq yang selaras. Versi yang dikompilasi dari Stringtie2 dapat diunduh dari https://ccb.jhu.edu/software/stringtie/#install.
Paket perangkat lunak bedtools diperlukan oleh GeneMark-ETP jika Anda ingin menjalankan Braker dengan data RNA-seq dan protein. Anda dapat mengunduh bedtools dari https://github.com/arq5x/bedtools2/releases. Di sini, Anda dapat mengunduh versi bedtools.static.binary , misalnya
wget https://github.com/arq5x/bedtools2/releases/download/v2.30.0/bedtools.static.binary
mv bedtools.static.binary bedtools
chmod a+x
atau Anda dapat mengunduh bedtools-2.30.0.tar.gz dan mengkompilasinya dari sumber menggunakan make , misalnya
wget https://github.com/arq5x/bedtools2/releases/download/v2.30.0/bedtools-2.30.0.tar.gz
tar -zxvf bedtools-2.30.0.tar.gz
cd bedtools2
make
Lihat https://bedtools.readthedocs.io/en/latest/content/installation.html untuk informasi lebih lanjut.
GFFREAD adalah perangkat lunak utilitas yang diperlukan oleh GeneMark-ETP. Ini dapat diunduh dari https://github.com/gpertea/gffread/releases/download/v0.12.7/gffread-0.12.7.linux_x86_64.tar.gz dan diinstal dengan make , eg
wget https://github.com/gpertea/gffread/releases/download/v0.12.7/gffread-0.12.7.Linux_x86_64.tar.gz
tar xzf gffread-0.12.7.Linux_x86_64.tar.gz
cd gffread-0.12.7.Linux_x86_64
make
Samtools tidak diperlukan untuk menjalankan Braker tanpa Genemark-ETP jika semua file Anda diformat, dengan benar (yaitu semua urutan harus memiliki nama fasta pendek dan unik). Jika Anda tidak yakin apakah semua file Anda diinstal dengan benar, mungkin bermanfaat untuk menginstal Samtools karena Braker dapat secara otomatis memperbaiki masalah format tertentu dengan menggunakan SamTools.
Sebagai prasyarat untuk Samtools, unduh dan instal htslib (misalnya git clone https://github.com/samtools/htslib.git , ikuti dokumentasi htslib untuk instalasi).
Unduh dan instal Samtools (misalnya git clone git://github.com/samtools/samtools.git ), selanjutnya ikuti dokumentasi Samtools untuk instalasi).
Jika sudah ada di variabel $PATH Anda, Braker akan menemukan Samtools, secara otomatis. Jika tidak, Braker dapat menemukan samtools baik dengan mengambil argumen baris perintah ( --SAMTOOLS_PATH=/your_path_to_samtools/ ), atau dengan menggunakan variabel lingkungan $SAMTOOLS_PATH . Untuk mengekspor variabel, misalnya untuk sesi bash Anda saat ini, ketik:
export SAMTOOLS_PATH=/your_path_to_samtools/
Tambahkan baris di atas ke skrip startup (misalnya ~/.bashrc ) untuk mengatur variabel lingkungan untuk semua sesi bash.
Jika Biopython diinstal, Braker dapat menghasilkan Fasta-Files dengan urutan pengkodean dan sekuens protein yang diprediksi oleh Augustus dan menghasilkan hub data trek untuk visualisasi Braker yang dijalankan dengan Makehub R16 . Ini adalah langkah opsional. Yang pertama dapat dinonaktifkan dengan bendera baris perintah --skipGetAnnoFromFasta , yang kedua dapat diaktifkan dengan menggunakan opsi baris perintah --makehub [email protected] , Biopython tidak diperlukan jika tidak satu pun dari langkah opsional ini ini akan dilakukan.
Di Ubuntu, instal Python3 Package Manager dengan:
`sudo apt-get install python3-pip`
Kemudian, pasang Biopython dengan:
`sudo pip3 install biopython`
CDBFASTA dan CDBYank diminta oleh Braker untuk mengoreksi gen Augustus dengan kodon stop frame (kodon berhenti yang disambung) menggunakan skrip Augustus fix_in_frame_stop_codon_genes.py. Ini dapat dilewati dengan --skip_fixing_broken_genes .
Di Ubuntu, instal CDBFASTA dengan:
sudo apt-get install cdbfasta
Untuk sistem lain, misalnya Anda dapat memperoleh CDBFASTA dari https://github.com/gpertea/cdbfasta, misalnya:
git clone https://github.com/gpertea/cdbfasta.git
cd cdbfasta
make all
Di Ubuntu, CDBFASTA dan CDBYank akan berada di variabel $PATH Anda setelah instalasi, dan Braker akan secara otomatis menemukannya. Namun, Anda memiliki opsi untuk menentukan lokasi biner cdbfasta dan cdbyank dengan dua cara lain:
$CDBTOOLS_PATH , misalnya dalam file ~/.bashrc Anda: export CDBTOOLS_PATH=/path/to/cdbtools/
--CDBTOOLS_PATH=/path/to/cdbtools/ to braker.pl . CATATAN: Dukungan spaln yang berdiri sendiri (ouside of prothint) di dalam Braker sudah usang.
Alat ini diperlukan jika Anda menjalankan Prothint atau jika Anda ingin menjalankan protein ke perataan genom dengan Braker menggunakan Spaln di luar Prothint. Menggunakan Spaln di luar Prothint adalah pendekatan yang cocok hanya jika spesies beranotasi jarak evolusi pendek ke genom target Anda tersedia. Kami merekomendasikan menjalankan Spaln melalui Prothint untuk Braker. Prothint membawa biner Spaln. Jika itu tidak berfungsi pada sistem Anda, unduh Spaln dari https://github.com/ogotoh/spaln. Buka pupuk dan instal sesuai dengan spaln/doc/SpalnReadMe22.pdf .
Braker akan mencoba menemukan Spaln yang dapat dieksekusi dengan menggunakan variabel lingkungan $ALIGNMENT_TOOL_PATH . Atau, ini dapat disediakan sebagai argumen baris perintah ( --ALIGNMENT_TOOL_PATH=/your/path/to/spaln ).
Alat ini hanya diperlukan jika Anda ingin menambahkan UTR (dari data RNA-seq) ke gen yang diprediksi atau jika Anda ingin melatih parameter UTR untuk Augustus dan memprediksi gen dengan UTR. Bagaimanapun, Gushr membutuhkan input data RNA-seq.
Gushr tersedia untuk diunduh di https://github.com/gaius-augustus/gushr. Dapatkan dengan mengetik:
git clone https://github.com/Gaius-Augustus/GUSHR.git
Gushr mengeksekusi file jar Gemoma R19, R20, R21 , dan file jar ini membutuhkan Java 1.8. Di Ubuntu, Anda dapat menginstal Java 1.8 dengan perintah berikut:
sudo apt-get install openjdk-8-jdk
Jika Anda memiliki beberapa versi java yang diinstal pada sistem Anda, pastikan Anda mengaktifkan 1.8 sebelum menjalankan Braker dengan Java dengan menjalankan
sudo update-alternatives --config java
dan memilih versi yang benar.
Jika Anda beralih --UTR=on , bamtowig.py akan memerlukan alat berikut yang dapat diunduh dari http://hgdownload.soe.ucsc.edu/admin/exe:
Twobitinfo
fatotwobit
Adalah opsional untuk menginstal alat ini ke jalur $ Anda. Jika tidak, dan Anda beralih --UTR=on , bamtowig.py akan secara otomatis mengunduhnya ke direktori yang berfungsi.
Jika Anda ingin Automaticaly menghasilkan hub data trek dari Braker Run Anda, perangkat lunak Makehub, tersedia di https://github.com/gaius-augustus/makehub diperlukan. Unduh Perangkat Lunak (baik dengan menjalankan git clone https://github.com/Gaius-Augustus/MakeHub.git , atau dengan memilih rilis dari https://github.com/gaius-ugustus/makehub/releases. Paket Jika Anda mengunduh rilis (misalnya unzip MakeHub.zip atau tar -zxvf MakeHub.tar.gz .
Braker akan mencoba menemukan skrip make_hub.py dengan menggunakan variabel lingkungan $MAKEHUB_PATH . Atau, ini dapat disuplai sebagai argumen baris perintah ( --MAKEHUB_PATH=/your/path/to/MakeHub/ ). Braker juga dapat mencoba menebak lokasi Makehub di sistem Anda.
Jika Anda ingin Braker mengunduh perpustakaan RNA-seq dari SRA NCBI, alat SRA diperlukan. Anda bisa mendapatkan versi yang dikompilasi dari alat SRA dari http://daehwankimlab.github.io/hisat2/download/#version-heisat2-221.
Braker akan mencoba menemukan binari yang dapat dieksekusi dari SRA Toolkit (FastQ-Dump, Prefetch) dengan menggunakan variabel lingkungan $SRATOOLS_PATH . Atau, ini dapat disediakan sebagai argumen baris perintah ( --SRATOOLS_PATH=/your/path/to/SRAToolkit/ ). Braker juga dapat mencoba menebak lokasi toolkit SRA di sistem Anda jika executable berada di variabel $PATH Anda.
Jika Anda ingin menggunakan RNA-seq yang tidak selaras dibaca, perangkat lunak HISAT2 diperlukan untuk memetakannya ke genom. Versi yang dikompilasi dari HISAT2 dapat diunduh dari http://daehwankimlab.github.io/hisat2/download/#version-heisat2-221.
Braker akan mencoba menemukan binari HISAT2 yang dapat dieksekusi (HISAT2, HISAT2-Build) dengan menggunakan variabel lingkungan $HISAT2_PATH . Atau, ini dapat disediakan sebagai argumen baris perintah ( --HISAT2_PATH=/your/path/to/HISAT2/ ). Braker juga dapat mencoba menebak lokasi HISAT2 di sistem Anda jika executable berada di variabel $PATH Anda.
Jika Anda ingin menjalankan Tsebra di dalam Braker dalam mode memaksimalkan kelengkapan Busco, Anda perlu menginstal Compleasm.
wget https://github.com/huangnengCSU/compleasm/releases/download/v0.2.4/compleasm-0.2.4_x64-linux.tar.bz2
tar -xvjf compleasm-0.2.4_x64-linux.tar.bz2 &&
Tambahkan folder yang dihasilkan compleasm_kit ke variabel $PATH Anda, misalnya:
export PATH=$PATH:/your/path/to/compleasm_kit
Compleasm membutuhkan panda, yang dapat diinstal dengan:
pip install pandas
Braker (Braker.pl) menggunakan GetConf untuk melihat berapa banyak utas yang dapat dijalankan pada sistem Anda. Di Ubuntu, Anda dapat menginstalnya dengan:
sudo apt-get install libc-bin
Berikut ini, kami menjelaskan "tipikal" Braker untuk tipe data input yang berbeda. Secara umum, kami menyarankan Anda menjalankan Braker pada urutan genomik yang telah diulang softmasked untuk pengulangan. Braker hanya boleh diterapkan pada genom yang telah diulang untuk diulang!
This approach is suitable for genomes of species for which RNA-Seq libraries with good transcriptome coverage are available and for which protein data is not at hand. The pipeline is illustrated in Figure 2.
BRAKER has several ways to receive RNA-Seq data as input:
You can provide ID(s) of RNA-Seq libraries from SRA (in case of multiple IDs, separate them by comma) as argument to --rnaseq_sets_ids . The libraries belonging to the IDs are then downloaded automatically by BRAKER, eg:
braker.pl --species=yourSpecies --genome=genome.fasta
--rnaseq_sets_ids=SRA_ID1,SRA_ID2
You can use local FASTQ file(s) of unaligned reads as input. In this case, you have to provide BRAKER with the ID(s) of the RNA-Seq set(s) as argument to --rnaseq_sets_ids and the path(s) to the directories, where the FASTQ files are located as argument to --rnaseq_sets_dirs . For each ID ID , BRAKER will search in these directories for one FASTQ file named ID.fastq if the reads are unpaired, or for two FASTQ files named ID_1.fastq and ID_2.fastq if they are paired.
For example, if you have a paired library called 'SRA_ID1' and an unpaired library named 'SRA_ID2', you have to have a directory /path/to/local/fastq/files/ , where the files SRA_ID1_1.fastq , SRA_ID1_2.fastq , and SRA_ID2.fastq reside. Then, you could run BRAKER with following command:
braker.pl --species=yourSpecies --genome=genome.fasta
--rnaseq_sets_ids=SRA_ID1,SRA_ID2
--rnaseq_sets_dirs=/path/to/local/fastq/files/
There are two ways of supplying BRAKER with RNA-Seq data as bam file(s). First, you can do it in the same way as you would supply FASTQ file(s): Provide the ID(s)/name(s) of your bam file(s) as argument to --rnaseq_sets_ids and specify directories where the bam files reside with --rnaseq_sets_dirs . BRAKER will automatically detect that these ID(s) are bam and not FASTQ file(s), eg:
braker.pl --species=yourSpecies --genome=genome.fasta
--rnaseq_sets_ids=BAM_ID1,BAM_ID2
--rnaseq_sets_dirs=/path/to/local/bam/files/
Second, you can specify the paths to your bam file(s) directly, eg can either extract RNA-Seq spliced alignment information from bam files, or it can use such extracted information, directly.
braker.pl --species=yourSpecies --genome=genome.fasta
--bam=file1.bam,file2.bam
Please note that we generally assume that bam files were generated with HiSat2 because that is the aligner that would also be executed by BRAKER3 with fastq input. If you want for some reason to generate the bam files with STAR, use the option --outSAMstrandField intronMotif of STAR to produce files that are compatible wiht StringTie in BRAKER3.
In order to run BRAKER with RNA-Seq spliced alignment information that has already been extracted, run:
braker.pl --species=yourSpecies --genome=genome.fasta
--hints=hints1.gff,hints2.gff
The format of such a hints file must be as follows (tabulator separated file):
chrName b2h intron 6591 8003 1 + . pri=4;src=E
chrName b2h intron 6136 9084 11 + . mult=11;pri=4;src=E
...
The source b2h in the second column and the source tag src=E in the last column are essential for BRAKER to determine whether a hint has been generated from RNA-Seq data.
It is also possible to provide RNA-Seq sets in different ways for the same BRAKER run, any combination of above options is possible. It is not recommended to provide RNA-Seq data with --hints if you run BRAKER in ETPmode (RNA-Seq and protein data), because GeneMark-ETP won't use these hints!
This approach is suitable for genomes of species for which no RNA-Seq libraries are available. A large database of proteins (with possibly longer evolutionary distance to the target species) should be used in this case. This mode is illustrated in figure 9.
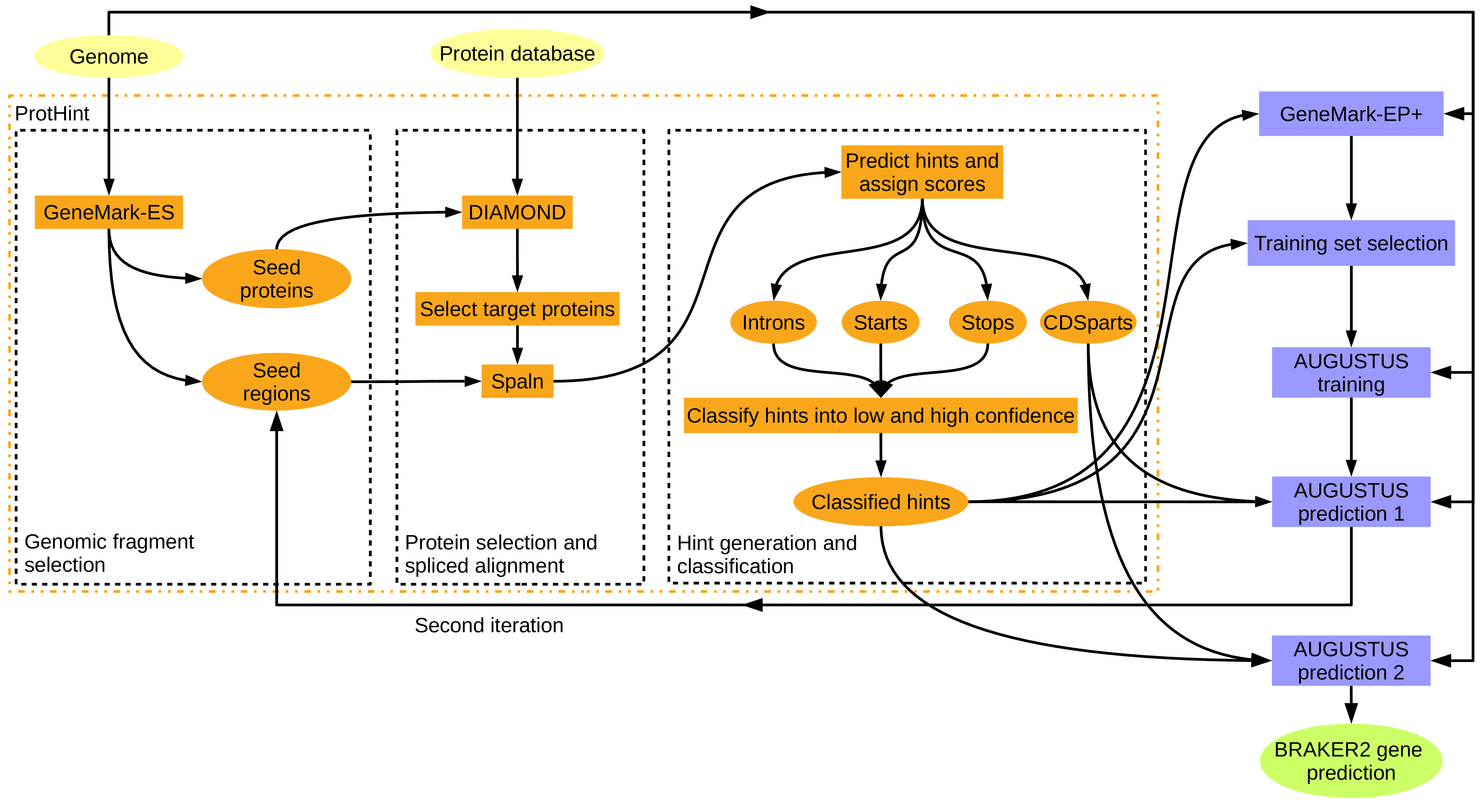
Figure 9: BRAKER with proteins of any evolutionary distance. ProtHint protein mapping pipelines is used to generate protein hints. ProtHint automatically determines which alignments are from close relatives, and which are from rather distant relatives.
For running BRAKER in this mode, type:
braker.pl --genome=genome.fa --prot_seq=proteins.fa
We recommend using OrthoDB as basis for proteins.fa . The instructions on how to prepare the input OrthoDB proteins are documented here: https://github.com/gatech-genemark/ProtHint#protein-database-preparation.
You can of course add additional protein sequences to that file, or try with a completely different database. Any database will need several representatives for each protein, though.
Instead of having BRAKER run ProtHint, you can also start BRAKER with hints already produced by ProtHint, by providing ProtHint's prothint_augustus.gff output:
braker.pl --genome=genome.fa --hints=prothint_augustus.gff
The format of prothint_augustus.gff in this mode looks like this:
2R ProtHint intron 11506230 11506648 4 + . src=M;mult=4;pri=4
2R ProtHint intron 9563406 9563473 1 + . grp=69004_0:001de1_702_g;src=C;pri=4;
2R ProtHint intron 8446312 8446371 1 + . grp=43151_0:001cae_473_g;src=C;pri=4;
2R ProtHint intron 8011796 8011865 2 - . src=P;mult=1;pri=4;al_score=0.12;
2R ProtHint start 234524 234526 1 + . src=P;mult=1;pri=4;al_score=0.08;
The prediction of all hints with src=M will be enforced. Hints with src=C are 'chained evidence', ie they will only be incorporated if all members of the group (grp=...) can be incorporated in a single transcript. All other hints have src=P in the last column. Supported features in column 3 are intron , start , stop and CDSpart .
If RNA-Seq (and only RNA-Seq) data is provided to BRAKER as a bam-file, and if the genome is softmasked for repeats, BRAKER can automatically train UTR parameters for AUGUSTUS. After successful training of UTR parameters, BRAKER will automatically predict genes including coverage information form RNA-Seq data. Example call:
braker.pl --species=yourSpecies --genome=genome.fasta
--bam=file.bam --UTR=on
Warnings:
This feature is experimental!
--UTR=on is currently not compatible with bamToWig.py as released in AUGUSTUS 3.3.3; it requires the current development code version from the github repository (git clone https://github.com/Gaius-Augustus/Augustus.git).
--UTR=on increases memory consumption of AUGUSTUS. Carefully monitor jobs if your machine was close to maxing RAM without --UTR=on! Reducing the number of cores will also reduce RAM consumption.
UTR prediction sometimes improves coding sequence prediction accuracy, but not always. If you try this feature, carefully compare results with and without UTR parameters, afterwards (eg in UCSC Genome Browser).
For running BRAKER without UTR parameters, it is not very important whether RNA-Seq data was generated by a stranded protocol (because spliced alignments are 'artificially stranded' by checking the splice site pattern). However, for UTR training and prediction, stranded libraries may provide information that is valuable for BRAKER.
After alignment of the stranded RNA-Seq libraries, separate the resulting bam file entries into two files: one for plus strand mappings, one for minus strand mappings. Call BRAKER as follows:
braker.pl --species=yourSpecies --genome=genome.fasta
--bam=plus.bam,minus.bam --stranded=+,-
--UTR=on
You may additionally include bam files from unstranded libraries. Those files will not used for generating UTR training examples, but they will be included in the final gene prediction step as unstranded coverage information, example call:
braker.pl --species=yourSpecies --genome=genome.fasta
--bam=plus.bam,minus.bam,unstranded.bam
--stranded=+,-,. --UTR=on
Warning: This feature is experimental and currently has low priority on our maintenance list!
The native mode for running BRAKER with RNA-Seq and protein data. This will call GeneMark-ETP, which will use RNA-Seq and protein hints for training GeneMark-ETP. Subsequently, AUGUSTUS is trained on 'high-confindent' genes (genes with very high extrinsic evidence support) from the GeneMark-ETP prediction and a set of genes is predicted by AUGUSTUS. In a last step, the predictions of AUGUSTUS and GeneMark-ETP are combined using TSEBRA.
Alignment of RNA-Seq reads
GeneMark-ETP utilizes Stringtie2 to assemble RNA-Seq data, which requires that the aligned reads (BAM files) contain the XS (strand) tag for spliced reads. Therefore, if you align your reads with HISAT2, you must enable the --dta option, or if you use STAR, you must use the --outSAMstrandField intronMotif option. TopHat alignments include this tag by default.
To call the pipeline in this mode, you have to provide it with a protein database using --prot_seq (as described in BRAKER with protein data), and RNA-Seq data either by their SRA ID so that they are downloaded by BRAKER, as unaligned reads in FASTQ format, and/or as aligned reads in bam format (as described in BRAKER with RNA-Seq data). You could also specify already processed extrinsic evidence using the --hints option. However, this is not recommend for a normal BRAKER run in ETPmode, as these hints won't be used in the GeneMark-ETP step. Only use --hints when you want to skip the GenMark-ETP step!
Examples of how you could run BRAKER in ETPmode:
braker.pl --genome=genome.fa --prot_seq=orthodb.fa
--rnaseq_sets_ids=SRA_ID1,SRA_ID2
--rnaseq_sets_dirs=/path/to/local/RNA-Seq/files/
braker.pl --genome=genome.fa --prot_seq=orthodb.fa
--rnaseq_sets_ids=SRA_ID1,SRA_ID2,SRA_ID3
braker.pl --genome=genome.fa --prot_seq=orthodb.fa
--bam=/path/to/SRA_ID1.bam,/path/to/SRA_ID2.bam
A preliminary protocol for integration of assembled subreads from PacBio ccs sequencing in combination with short read Illumina RNA-Seq and protein database is described at https://github.com/Gaius-Augustus/BRAKER/blob/master/docs/long_reads/long_read_protocol.md
We forked GeneMark-ETP and hard coded that StringTie will perform long read assembly in that particular version. If you want to use this 'fast-hack' version for BRAKER, you have to prepare the BAM file with long read to genome spliced alignments outside of BRAKER, eg:
T=48 # adapt to your number of threads
minimap2 -t${T} -ax splice:hq -uf genome.fa isoseq.fa > isoseq.sam
samtools view -bS --threads ${T} isoseq.sam -o isoseq.bam
Pull the adapted container:
singularity build braker3_lr.sif docker://teambraker/braker3:isoseq
Calling BRAKER3 with a BAM file of spliced-aligned IsoSeq Reads:
singularity exec -B ${PWD}:${PWD} braker3_lr.sif braker.pl --genome=genome.fa --prot_seq=protein_db.fa –-bam=isoseq.bam --threads=${T}
Warning Do NOT mix short read and long read data in this BRAKER/GeneMark-ETP variant!
Warning The accuracy of gene prediction here heavily depends on the depth of your isoseq data. We verified with PacBio HiFi reads from 2022 that given sufficient completeness of the assembled transcriptome you will reach similar results as with short reads. However, we also observed a drop in accuracy compared to short reads when using other long read data sets with higher error rates and less sequencing depth.
Please run braker.pl --help to obtain a full list of options.
Compute AUGUSTUS ab initio predictions in addition to AUGUSTUS predictions with hints (additional output files: augustus.ab_initio.* . This may be useful for estimating the quality of training gene parameters when inspecting predictions in a Browser.
One or several command line arguments to be passed to AUGUSTUS, if several arguments are given, separate them by whitespace, ie "--first_arg=sth --second_arg=sth" . This may be be useful if you know that gene prediction in your particular species benefits from a particular AUGUSTUS argument during the prediction step.
Specifies the maximum number of threads that can be used during computation. BRAKER has to run some steps on a single thread, others can take advantage of multiple threads. If you use more than 8 threads, this will not speed up all parallelized steps, in particular, the time consuming optimize_augustus.pl will not use more than 8 threads. However, if you don't mind some threads being idle, using more than 8 threads will speed up other steps.
GeneMark-ETP option: run algorithm with branch point model. Use this option if you genome is a fungus.
Use the present config and parameter files if they exist for 'species'; will overwrite original parameters if BRAKER performs an AUGUSTUS training.
Execute CRF training for AUGUSTUS; resulting parameters are only kept for final predictions if they show higher accuracy than HMM parameters. This increases runtime!
Change the parameter
Generate UTR training examples for AUGUSTUS from RNA-Seq coverage information, train AUGUSTUS UTR parameters and predict genes with AUGUSTUS and UTRs, including coverage information for RNA-Seq as evidence. This is an experimental feature!
If you performed a BRAKER run without --UTR=on, you can add UTR parameter training and gene prediction with UTR parameters (and only RNA-Seq hints) with the following command:
braker.pl --genome=../genome.fa --addUTR=on
--bam=../RNAseq.bam --workingdir=$wd
--AUGUSTUS_hints_preds=augustus.hints.gtf
--threads=8 --skipAllTraining --species=somespecies
Modify augustus.hints.gtf to point to the AUGUSTUS predictions with hints from previous BRAKER run; modify flaning_DNA value to the flanking region from the log file of your previous BRAKER run; modify some_new_working_directory to the location where BRAKER should store results of the additional BRAKER run; modify somespecies to the species name used in your previous BRAKER run.
Add UTRs from RNA-Seq converage information to AUGUSTUS gene predictions using GUSHR. No training of UTR parameters and no gene prediction with UTR parameters is performed.
If you performed a BRAKER run without --addUTR=on, you can add UTRs results of a previous BRAKER run with the following command:
braker.pl --genome=../genome.fa --addUTR=on
--bam=../RNAseq.bam --workingdir=$wd
--AUGUSTUS_hints_preds=augustus.hints.gtf --threads=8
--skipAllTraining --species=somespecies
Modify augustus.hints.gtf to point to the AUGUSTUS predictions with hints from previous BRAKER run; modify some_new_working_directory to the location where BRAKER should store results of the additional BRAKER run; this run will not modify AUGUSTUS parameters. We recommend that you specify the original species of the original run with --species=somespecies . Otherwise, BRAKER will create an unneeded species parameters directory Sp_* .
If --UTR=on is enabled, strand-separated bam-files can be provided with --bam=plus.bam,minus.bam . In that case, --stranded=... should hold the strands of the bam files ( + for plus strand, - for minus strand, . for unstranded). Note that unstranded data will be used in the gene prediction step, only, if the parameter --stranded=... is set. This is an experimental feature! GUSHR currently does not take advantage of stranded data.
If --makehub and [email protected] (with your valid e-mail adress) are provided, a track data hub for visualizing results with the UCSC Genome Browser will be generated using MakeHub (https://github.com/Gaius-Augustus/MakeHub).
By default, GeneMark-ES/ET/EP/ETP uses a probability of 0.001 for predicting the donor splice site pattern GC (instead of GT). It may make sense to increase this value for species where this donor splice site is more common. For example, in the species Emiliania huxleyi , about 50% of donor splice sites have the pattern GC (https://media.nature.com/original/nature-assets/nature/journal/v499/n7457/extref/nature12221-s2.pdf, page 5).
Use a species-specific lineage, eg arthropoda_odb10 for an arthropod. BRAKER does not support auto-typing of the lineage.
Specifying a BUSCO-lineage invokes two changes in BRAKER R28 :
BRAKER will run compleasm with the specified lineage in genome mode and convert the detected BUSCO matches into hints for AUGUSTUS. This may increase the number of BUSCOs in the augustus.hints.gtf file slightly.
BRAKER will invoke best_by_compleasm.py to check whether the braker.gtf file that is by default generated by TSEBRA has the lowest amount of missing BUSCOs compared to the augustus.hints.gtf and the genemark.gtf file. If not, the following decision schema is applied to re-run TSEBRA to minimize the missing BUSCOs in the final output of BRAKER (always braker.gtf). If an alternative and better gene set is created, the original braker.gtf gene set is moved to a directory called braker_original. Information on what happened during the best_by_compleasm.py run is written to the file best_by_compleasm.log.
![best_by_busco[fig14]](https://images.downcodes.com/uploads/20250214/img_67aee79a11fd439.png)
Please note that using BUSCO to assess the quality of a gene set, in particular when comparing BRAKER to other pipelines, does not make sense once you specified a BUSCO lineage. We recommend that you use other measures to assess the quality of your gene set, eg by comparing it to a reference gene set or running OMArk.
BRAKER produces several important output files in the working directory.
braker.gtf: Final gene set of BRAKER. This file may contain different contents depending on how you called BRAKER
in ETPmode: Final gene set of BRAKER consisting of genes predicted by AUGUSTUS and GeneMark-ETP that were combined and filtered by TSEBRA.
otherwise: Union of augustus.hints.gtf and reliable GeneMark-ES/ET/EP predictions (genes fully supported by external evidence). In --esmode , this is the union of augustus.ab_initio.gtf and all GeneMark-ES genes. Thus, this set is generally more sensitive (more genes correctly predicted) and can be less specific (more false-positive predictions can be present). This output is not necessarily better than augustus.hints.gtf, and it is not recommended to use it if BRAKER was run in ESmode.
braker.codingseq: Final gene set with coding sequences in FASTA format
braker.aa: Final gene set with protein sequences in FASTA format
braker.gff3: Final gene set in gff3 format (only produced if the flag --gff3 was specified to BRAKER.
Augustus/*: Augustus gene set(s) in as gtf/conding/aa files
GeneMark-E*/genemark.gtf: Genes predicted by GeneMark-ES/ET/EP/EP+/ETP in GTF-format.
hintsfile.gff: The extrinsic evidence data extracted from RNAseq.bam and/or protein data.
braker_original/*: Genes predicted by BRAKER (TSEBRA merge) before compleasm was used to improve BUSCO completeness
bbc/*: output folder of best_by_compleasm.py script from TSEBRA that is used to improve BUSCO completeness in the final output of BRAKER
Output files may be present with the following name endings and formats:
Coding sequences in FASTA-format are produced if the flag --skipGetAnnoFromFasta was not set.
Protein sequence files in FASTA-format are produced if the flag --skipGetAnnoFromFasta was not set.
For details about gtf format, see http://www.sanger.ac.uk/Software/formats/GFF/. A GTF-format file contains one line per predicted exon. Contoh:
HS04636 AUGUSTUS initial 966 1017 . + 0 transcript_id "g1.1"; gene_id "g1";
HS04636 AUGUSTUS internal 1818 1934 . + 2 transcript_id "g1.1"; gene_id "g1";
The columns (fields) contain:
seqname source feature start end score strand frame transcript ID and gene ID
If the --makehub option was used and MakeHub is available on your system, a hub directory beginning with the name hub_ will be created. Copy this directory to a publicly accessible web server. A file hub.txt resides in the directory. Provide the link to that file to the UCSC Genome Browser for visualizing results.
An incomplete example data set is contained in the directory BRAKER/example . In order to complete the data set, please download the RNA-Seq alignment file (134 MB) with wget :
cd BRAKER/example
wget http://topaz.gatech.edu/GeneMark/Braker/RNAseq.bam
In case you have trouble accessing that file, there's also a copy available from another server:
cd BRAKER/example
wget http://bioinf.uni-greifswald.de/augustus/datasets/RNAseq.bam
The example data set was not compiled in order to achieve optimal prediction accuracy, but in order to quickly test pipeline components. The small subset of the genome used in these test examples is not long enough for BRAKER training to work well.
Data corresponds to the last 1,000,000 nucleotides of Arabidopsis thaliana 's chromosome Chr5, split into 8 artificial contigs.
RNA-Seq alignments were obtained by VARUS.
The protein sequences are a subset of OrthoDB v10 plants proteins.
List of files:
genome.fa - genome file in fasta formatRNAseq.bam - RNA-Seq alignment file in bam format (this file is not a part of this repository, it must be downloaded separately from http://topaz.gatech.edu/GeneMark/Braker/RNAseq.bam)RNAseq.hints - RNA-Seq hints (can be used instead of RNAseq.bam as RNA-Seq input to BRAKER)proteins.fa - protein sequences in fasta formatThe below given commands assume that you configured all paths to tools by exporting bash variables or that you have the necessary tools in your $PATH.
The example data set also contains scripts tests/test*.sh that will execute below listed commands for testing BRAKER with the example data set. You find example results of AUGUSTUS and GeneMark-ES/ET/EP/ETP in the folder results/test* . Be aware that BRAKER contains several parts where random variables are used, ie results that you obtain when running the tests may not be exactly identical. To compare your test results with the reference ones, you can use the compare_intervals_exact.pl script as follows:
# Compare CDS features
compare_intervals_exact.pl --f1 augustus.hints.gtf --f2 ../../results/test${N}/augustus.hints.gtf --verbose
# Compare transcripts
compare_intervals_exact.pl --f1 augustus.hints.gtf --f2 ../../results/test${N}/augustus.hints.gtf --trans --verbose
Several tests use --gm_max_intergenic 10000 option to make the test runs faster. It is not recommended to use this option in real BRAKER runs, the speed increase achieved by adjusting this option is negligible on full-sized genomes.
We give runtime estimations derived from computing on Intel(R) Xeon(R) CPU E5530 @ 2.40GHz .
The following command will run the pipeline according to Figure 3:
braker.pl --genome genome.fa --bam RNAseq.bam --threads N --busco_lineage=lineage_odb10
This test is implemented in test1.sh , expected runtime is ~20 minutes.
The following command will run the pipeline according to Figure 4:
braker.pl --genome genome.fa --prot_seq proteins.fa --threads N --busco_lineage=lineage_odb10
This test is implemented in test2.sh , expected runtime is ~20 minutes.
The following command will run a pipeline that first trains GeneMark-ETP with protein and RNA-Seq hints and subsequently trains AUGUSTUS on the basis of GeneMark-ETP predictions. AUGUSTUS predictions are also performed with hints from both sources, see Figure 5.
Run with local RNA-Seq file:
braker.pl --genome genome.fa --prot_seq proteins.fa --bam ../RNAseq.bam --threads N --busco_lineage=lineage_odb10
This test is implemented in test3.sh , expected runtime is ~20 minutes.
Download RNA-Seq library from Sequence Read Archive (~1gb):
braker.pl --genome genome.fa --prot_seq proteins.fa --rnaseq_sets_ids ERR5767212 --threads N --busco_lineage=lineage_odb10
This test is implemented in test3_4.sh , expected runtime is ~35 minutes.
The training step of all pipelines can be skipped with the option --skipAllTraining . This means, only AUGUSTUS predictions will be performed, using pre-trained, already existing parameters. For example, you can predict genes with the command:
braker.pl --genome=genome.fa --bam RNAseq.bam --species=arabidopsis
--skipAllTraining --threads N
This test is implemented in test4.sh , expected runtime is ~1 minute.
The following command will run the pipeline with no extrinsic evidence:
braker.pl --genome=genome.fa --esmode --threads N
This test is implemented in test5.sh , expected runtime is ~20 minutes.
The following command will run BRAKER with training UTR parameters from RNA-Seq coverage data:
braker.pl --genome genome.fa --bam RNAseq.bam --UTR=on --threads N
This test is implemented in test6.sh , expected runtime is ~20 minutes.
The following command will add UTRs to augustus.hints.gtf from RNA-Seq coverage data:
braker.pl --genome genome.fa --bam RNAseq.bam --addUTR=on --threads N
This test is implemented in test7.sh , expected runtime is ~20 minutes.
There is currently no clean way to restart a failed BRAKER run (after solving some problem). However, it is possible to start a new BRAKER run based on results from a previous run -- given that the old run produced the required intermediate results. We will in the following refer to the old working directory with variable ${BRAKER_OLD} , and to the new BRAKER working directory with ${BRAKER_NEW} . The file what-to-cite.txt will always only refer to the software that was actually called by a particular run. You might have to combine the contents of ${BRAKER_NEW}/what-to-cite.txt with ${BRAKER_OLD}/what-to-cite.txt for preparing a publication. The following figure illustrates at which points BRAKER run may be intercepted.
![braker-intercept[fig8]](https://images.downcodes.com/uploads/20250214/img_67aee79a12cab310.png)
Figure 10: Points for intercepting a BRAKER run and reusing intermediate results in a new BRAKER run.
This option is only possible for BRAKER in ETmode or EPmode and bukan in ETPmode!
If you have access to an existing BRAKER output that contains hintsfiles that were generated from extrinsic data, such as RNA-Seq or protein sequences, you can recycle these hints files in a new BRAKER run. Also, hints from a separate ProtHint run can be directly used in BRAKER.
The hints can be given to BRAKER with --hints ${BRAKER_OLD}/hintsfile.gff option. This is illustrated in the test files test1_restart1.sh , test2_restart1.sh , test4_restart1.sh . The other modes (for which this test is missing) cannot be restarted in this way.
The GeneMark result can be given to BRAKER with --geneMarkGtf ${BRAKER_OLD}/GeneMark*/genemark.gtf option if BRAKER is run in ETmode or EPmode. This is illustrated in the test files test1_restart2.sh , test2_restart2.sh , test5_restart2.sh .
In ETPmode, you can either provide BRAKER with the results of the GeneMarkETP step manually, with --geneMarkGtf ${BRAKER_OLD}/GeneMark-ETP/proteins.fa/genemark.gtf , --traingenes ${BRAKER_OLD}/GeneMark-ETP/training.gtf , and --hints ${BRAKER_OLD}/hintsfile.gff (see test3_restart1.sh for an example), or you can specify the previous GeneMark-ETP results with the option --gmetp_results_dir ${BRAKER_OLD}/GeneMark-ETP/ so that BRAKER can search for the files automatically (see test3_restart2.sh for an example).
The trained species parameters for AGUSTUS can be passed with --skipAllTraining and --species $speciesName options. This is illustrated in test*_restart3.sh files. Note that in ETPmode you have to specify the GeneMark files as described in Option 2!
Before reporting bugs, please check that you are using the most recent versions of GeneMark-ES/ET/EP/ETP, AUGUSTUS and BRAKER. Also, check the list of Common problems, and the Issue list on GitHub before reporting bugs. We do monitor open issues on GitHub. Sometimes, we are unable to help you, immediately, but we try hard to solve your problems.
If you found a bug, please open an issue at https://github.com/Gaius-Augustus/BRAKER/issues (or contact [email protected] or [email protected]).
Information worth mentioning in your bug report:
Check in braker/yourSpecies/braker.log at which step braker.pl crashed.
There are a number of other files that might be of interest, depending on where in the pipeline the problem occurred. Some of the following files will not be present if they did not contain any errors.
braker/yourSpecies/errors/bam2hints.*.stderr - will give details on a bam2hints crash (step for converting bam file to intron gff file)
braker/yourSpecies/hintsfile.gff - is this file empty? If yes, something went wrong during hints generation - does this file contain hints from source “b2h” and of type “intron”? If not: GeneMark-ET will not be able to execute properly. Conversely, GeneMark-EP+ will not be able to execute correctly if hints from the source "ProtHint" are missing.
braker/yourSpecies/spaln/*err - errors reported by spaln
braker/yourSpecies/errors/GeneMark-{ET,EP,ETP}.stderr - errors reported by GeneMark-ET/EP+/ETP
braker/yourSpecies/errors/GeneMark-{ET,EP,ETP).stdout - may give clues about the point at which errors in GeneMark-ET/EP+/ETP occured
braker/yourSpecies/GeneMark-{ET,EP,ETP}/genemark.gtf - is this file empty? If yes, something went wrong during executing GeneMark-ET/EP+/ETP
braker/yourSpecies/GeneMark-{ET,EP}/genemark.f.good.gtf - is this file empty? If yes, something went wrong during filtering GeneMark-ET/EP+ genes for training AUGUSTUS
braker/yourSpecies/genbank.good.gb - try a “grep -c LOCUS genbank.good.gb” to determine the number of training genes for training AUGUSTUS, should not be low
braker/yourSpecies/errors/firstetraining.stderr - contains errors from first iteration of training AUGUSTUS
braker/yourSpecies/errors/secondetraining.stderr - contains errors from second iteration of training AUGUSTUS
braker/yourSpecies/errors/optimize_augustus.stderr - contains errors optimize_augustus.pl (additional training set for AUGUSTUS)
braker/yourSpecies/errors/augustus*.stderr - contain AUGUSTUS execution errors
braker/yourSpecies/startAlign.stderr - if you provided a protein fasta file, something went wrong during protein alignment
braker/yourSpecies/startAlign.stdout - may give clues on at which point protein alignment went wrong
BRAKER complains that the RNA-Seq file does not correspond to the provided genome file, but I am sure the files correspond to each other!
Please check the headers of the genome FASTA file. If the headers are long and contain whitespaces, some RNA-Seq alignment tools will truncate sequence names in the BAM file. This leads to an error with BRAKER. Solution: shorten/simplify FASTA headers in the genome file before running the RNA-Seq alignment and BRAKER.
GeneMark fails!
(a) GeneMark by default only uses contigs longer than 50k for training. If you have a highly fragmented assembly, this might lead to "no data" for training. You can override the default minimal length by setting the BRAKER argument --min_contig=10000 .
(b) see "[something] failed to execute" below.
[something] failed to execute!
When providing paths to software to BRAKER, please use absolute, non-abbreviated paths. For example, BRAKER might have problems with --SAMTOOLS_PATH=./samtools/ or --SAMTOOLS_PATH=~/samtools/ . Please use SAMTOOLS_PATH=/full/absolute/path/to/samtools/ , instead. This applies to all path specifications as command line options to braker.pl . Relative paths and absolute paths will not pose problems if you export a bash variable, instead, or if you append the location of tools to your $PATH variable.
GeneMark-ETP in BRAKER dies with '/scratch/11232323': No such file or directory.
This appears to be related to sorting large files, and it's a system configuration depending problem. Solve it with export TMPDIR=/tmp/ before calling BRAKER via Singularity.
BRAKER cannot find the Augustus script XYZ...
Update Augustus from github with git clone https://github.com/Gaius-Augustus/Augustus.git . Do not use Augustus from other sources. BRAKER is highly dependent on an up-to-date Augustus. Augustus releases happen rather rarely, updates to the Augustus scripts folder occur rather frequently.
Does BRAKER depend on Python3?
It does. The python scripts employed by BRAKER are not compatible with Python2.
Why does BRAKER predict more genes than I expected?
If transposable elements (or similar) have not been masked appropriately, AUGUSTUS tends to predict those elements as protein coding genes. This can lead to a huge number genes. You can check whether this is the case for your project by BLASTing (or DIAMONDing) the predicted protein sequences against themselves (all vs. all) and counting how many of the proteins have a high number of high quality matches. You can use the output of this analysis to divide your gene set into two groups: the protein coding genes that you want to find and the repetitive elements that were additionally predicted.
I am running BRAKER in Anaconda and something fails...
Update AUGUSTUS and BRAKER from github with git clone https://github.com/Gaius-Augustus/Augustus.git and git clone https://github.com/Gaius-Augustus/BRAKER.git . The Anaconda installation is great, but it relies on releases of AUGUSTUS and BRAKER - which are often lagging behind. Please use the current GitHub code, instead.
Why and where is the GenomeThreader support gone?
BRAKER is a joint project between teams from University of Greifswald and Georgia Tech. While the group of Mark Bordovsky from Georgia Tech contributes GeneMark expertise, the group of Mario Stanke from University of Greifswald contributes AUGUSTUS expertise. Using GenomeThreader to build training genes for AUGUSTUS in BRAKER circumvents execution of GeneMark. Thus, the GenomeThreader mode is strictly speaking not part of the BRAKER project. The previous functionality of BRAKER with GenomeThreader has been moved to GALBA at https://github.com/Gaius-Augustus/GALBA. Note that GALBA has also undergone extension for using Miniprot instead of GenomeThreader.
My BRAKER gene set has too many BUSCO duplicates!
AUGUSTUS within BRAKER can predict alternative splicing isoforms. Also the merge of the AUGUSTUS and GeneMark gene set by TSEBRA within BRAKER may result in additional isoforms for a single gene. The BUSCO duplicates usually come from alternative splicing isoforms, ie they are expected.
Augustus and/or etraining within BRAKER complain that the file aug_cmdln_parameters.json is missing. Even though I am using the latest Singularity container!
BRAKER copies the AUGUSTUS_CONFIG_PATH folder to a writable location. In older versions of Augustus, that file was indeed not existing. If the local writable copy of a folder already exists, BRAKER will not re-copy it. Simply delete the old folder. (It is often ~/.augustus , so you can simply do rm -rf ~/.augustus ; the folder might be residing in $PWD if your home directory was not writable).
I sit behind a firewall, compleasm cannot download the BUSCO files, what can I do? See Issue #785 (comment)
Since BRAKER is a pipeline that calls several Bioinformatics tools, publication of results obtained by BRAKER requires that not only BRAKER is cited, but also the tools that are called by BRAKER. BRAKER will output a file what-to-cite.txt in the BRAKER working directory, informing you about which exact sources apply to your run.
Always cite:
Stanke, M., Diekhans, M., Baertsch, R. and Haussler, D. (2008). Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics, doi: 10.1093/bioinformatics/btn013.
Stanke. M., Schöffmann, O., Morgenstern, B. and Waack, S. (2006). Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics 7, 62.
If you provided any kind of evidence for BRAKER, cite:
If you provided both short read RNA-Seq evidence and a large database of proteins, cite:
Gabriel, L., Bruna, T., Hoff, KJ, Ebel, M., Lomsadze, A., Borodovsky, M., Stanke, M. (2023). BRAKER3: Fully Automated Genome Annotation Using RNA-Seq and Protein Evidence with GeneMark-ETP, AUGUSTUS and TSEBRA. bioRxiV, doi: 10.1101/2023.06.10.54444910.1101/2023.01.01.474747.
Bruna, T., Lomsadze, A., Borodovsky, M. (2023). GeneMark-ETP: Automatic Gene Finding in Eukaryotic Genomes in Consistence with Extrinsic Data. bioRxiv, doi: 10.1101/2023.01.13.524024.
Kovaka, S., Zimin, AV, Pertea, GM, Razaghi, R., Salzberg, SL, & Pertea, M. (2019). Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome biology, 20(1):1-13.
Pertea, G., & Pertea, M. (2020). GFF utilities: GffRead and GffCompare. F1000Research, 9.
Quinlan, AR (2014). BEDTools: the Swiss‐army tool for genome feature analysis. Current protocols in bioinformatics, 47(1):11-12.
If the only source of evidence for BRAKER was a large database of protein sequences, cite:
If the only source of evidence for BRAKER was RNA-Seq data, cite:
Hoff, KJ, Lange, S., Lomsadze, A., Borodovsky, M. and Stanke, M. (2016). BRAKER1: unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics, 32(5):767-769.
Lomsadze, A., Paul DB, and Mark B. (2014) Integration of Mapped Rna-Seq Reads into Automatic Training of Eukaryotic Gene Finding Algorithm. Nucleic Acids Research 42(15): e119--e119
If you called BRAKER3 with an IsoSeq BAM file, or if you envoked the --busco_lineage option, cite:
If you called BRAKER with the --busco_lineage option, in addition, cite:
Simão, FA, Waterhouse, RM, Ioannidis, P., Kriventseva, EV, & Zdobnov, EM (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics, 31(19), 3210-3212.
Li, H. (2023). Protein-to-genome alignment with miniprot. Bioinformatics, 39(1), btad014.
Huang, N., & Li, H. (2023). compleasm: a faster and more accurate reimplementation of BUSCO. Bioinformatics, 39(10), btad595.
If any kind of AUGUSTUS training was performed by BRAKER, check carefully whether you configured BRAKER to use NCBI BLAST or DIAMOND. One of them was used to filter out redundant training gene structures.
If you used NCBI BLAST, please cite:
Altschul, AF, Gish, W., Miller, W., Myers, EW and Lipman, DJ (1990). A basic local alignment search tool. J Mol Biol 215:403--410.
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., and Madden, TL (2009). Blast+: architecture and applications. BMC bioinformatics, 10(1):421.
If you used DIAMOND, please cite:
If BRAKER was executed with a genome file and no extrinsic evidence, cite, then GeneMark-ES was used, cite:
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, YO and Borodovsky, M. (2005). Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Research, 33(20):6494--6506.
Ter-Hovhannisyan, V., Lomsadze, A., Chernoff, YO and Borodovsky, M. (2008). Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome research, pages gr--081612, 2008.
Hoff, KJ, Lomsadze, A., Borodovsky, M. and Stanke, M. (2019). Whole-Genome Annotation with BRAKER. Methods Mol Biol. 1962:65-95, doi: 10.1007/978-1-4939-9173-0_5.
If BRAKER was run with proteins as source of evidence, please cite all tools that are used by the ProtHint pipeline to generate hints:
Bruna, T., Lomsadze, A., & Borodovsky, M. (2020). GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genomics and Bioinformatics, 2(2), lqaa026.
Buchfink, B., Xie, C., Huson, DH (2015). Fast and sensitive protein alignment using DIAMOND. Nature Methods 12:59-60.
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, YO and Borodovsky, M. (2005). Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Research, 33(20):6494--6506.
Iwata, H., and Gotoh, O. (2012). Benchmarking spliced alignment programs including Spaln2, an extended version of Spaln that incorporates additional species-specific features. Nucleic acids research, 40(20), e161-e161.
Gotoh, O., Morita, M., Nelson, DR (2014). Assessment and refinement of eukaryotic gene structure prediction with gene-structure-aware multiple protein sequence alignment. BMC bioinformatics, 15(1), 189.
If BRAKER was executed with RNA-Seq alignments in bam-format, then SAMtools was used, cite:
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R.; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25(16):2078-9.
Barnett, DW, Garrison, EK, Quinlan, AR, Strömberg, MP and Marth GT (2011). BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics, 27(12):1691-2
If BRAKER downloaded RNA-Seq libraries from SRA using their IDs, cite SRA, SRA toolkit, and HISAT2:
Leinonen, R., Sugawara, H., Shumway, M., & International Nucleotide Sequence Database Collaboration. (2010). The sequence read archive. Nucleic acids research, 39(suppl_1), D19-D21.
SRA Toolkit Development Team (2020). SRA Toolkit. https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software.
Kim, D., Paggi, JM, Park, C., Bennett, C., & Salzberg, SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature biotechnology, 37(8):907-915.
If BRAKER was executed using RNA-Seq data in FASTQ format, cite HISAT2:
If BRAKER called MakeHub for creating a track data hub for visualization of BRAKER results with the UCSC Genome Browser, cite:
If BRAKER called GUSHR for generating UTRs, cite:
Keilwagen, J., Hartung, F., Grau, J. (2019) GeMoMa: Homology-based gene prediction utilizing intron position conservation and RNA-seq data. Methods Mol Biol. 1962:161-177, doi: 10.1007/978-1-4939-9173-0_9.
Keilwagen, J., Wenk, M., Erickson, JL, Schattat, MH, Grau, J., Hartung F. (2016) Using intron position conservation for homology-based gene prediction. Nucleic Acids Research, 44(9):e89.
Keilwagen, J., Hartung, F., Paulini, M., Twardziok, SO, Grau, J. (2018) Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinformatics, 19(1):189.
All source code, ie scripts/*.pl or scripts/*.py are under the Artistic License (see http://www.opensource.org/licenses/artistic-license.php).
[F1] EX = ES/ET/EP/ETP, all available for download under the name GeneMark-ES/ET/EP ↩
[F2] Please use the latest version from the master branch of AUGUSTUS distributed by the original developers, it is available from github at https://github.com/Gaius-Augustus/Augustus. Problems have been reported from users that tried to run BRAKER with AUGUSTUS releases maintained by third parties, ie Bioconda. ↩
[F4] install with sudo apt-get install cpanminus ↩
[F6] The binary may eg reside in bamtools/build/src/toolkit ↩
[R0] Bruna, Tomas, Hoff, Katharina J., Lomsadze, Alexandre, Stanke, Mario, and Borodovsky, Mark. 2021. “BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database." NAR Genomics and Bioinformatics 3(1):lqaa108.↩
[R1] Hoff, Katharina J, Simone Lange, Alexandre Lomsadze, Mark Borodovsky, and Mario Stanke. 2015. “BRAKER1: Unsupervised Rna-Seq-Based Genome Annotation with Genemark-et and Augustus.” Bioinformatics 32 (5). Oxford University Press: 767--69.↩
[R2] Lomsadze, Alexandre, Paul D Burns, and Mark Borodovsky. 2014. “Integration of Mapped Rna-Seq Reads into Automatic Training of Eukaryotic Gene Finding Algorithm.” Nucleic Acids Research 42 (15). Oxford University Press: e119--e119.↩
[R3] Stanke, Mario, Mark Diekhans, Robert Baertsch, and David Haussler. 2008. “Using Native and Syntenically Mapped cDNA Alignments to Improve de Novo Gene Finding.” Bioinformatics 24 (5). Oxford University Press: 637--44.↩
[R4] Stanke, Mario, Oliver Schöffmann, Burkhard Morgenstern, and Stephan Waack. 2006. “Gene Prediction in Eukaryotes with a Generalized Hidden Markov Model That Uses Hints from External Sources.” BMC Bioinformatics 7 (1). BioMed Central: 62.↩
[R5] Barnett, Derek W, Erik K Garrison, Aaron R Quinlan, Michael P Strömberg, and Gabor T Marth. 2011. “BamTools: A C++ Api and Toolkit for Analyzing and Managing Bam Files.” Bioinformatics 27 (12). Oxford University Press: 1691--2.↩
[R6] Li, Heng, Handsaker, Bob, Alec Wysoker, Tim Fennell, Jue Ruan, Nils Homer, Gabor Marth, Goncalo Abecasis, and Richard Durbin. 2009. “The Sequence Alignment/Map Format and Samtools.” Bioinformatics 25 (16). Oxford University Press: 2078--9.↩
[R7] Gremme, G. 2013. “Computational Gene Structure Prediction.” PhD thesis, Universität Hamburg.↩
[R8] Gotoh, Osamu. 2008a. “A Space-Efficient and Accurate Method for Mapping and Aligning cDNA Sequences onto Genomic Sequence.” Nucleic Acids Research 36 (8). Oxford University Press: 2630--8.↩
[R9] Iwata, Hiroaki, and Osamu Gotoh. 2012. “Benchmarking Spliced Alignment Programs Including Spaln2, an Extended Version of Spaln That Incorporates Additional Species-Specific Features.” Nucleic Acids Research 40 (20). Oxford University Press: e161--e161.↩
[R10] Osamu Gotoh. 2008b. “Direct Mapping and Alignment of Protein Sequences onto Genomic Sequence.” Bioinformatics 24 (21). Oxford University Press: 2438--44.↩
[R11] Slater, Guy St C, and Ewan Birney. 2005. “Automated Generation of Heuristics for Biological Sequence Comparison.” BMC Bioinformatics 6(1). BioMed Central: 31.↩
[R12] Altschul, SF, W. Gish, W. Miller, EW Myers, and DJ Lipman. 1990. “Basic Local Alignment Search Tool.” Journal of Molecular Biology 215:403--10.↩
[R13] Camacho, Christiam, et al. 2009. “BLAST+: architecture and applications.“ BMC Bioinformatics 1(1): 421.↩
[R14] Lomsadze, A., V. Ter-Hovhannisyan, YO Chernoff, and M. Borodovsky. 2005. “Gene identification in novel eukaryotic genomes by self-training algorithm.” Nucleic Acids Research 33 (20): 6494--6506. doi:10.1093/nar/gki937.↩
[R15] Ter-Hovhannisyan, Vardges, Alexandre Lomsadze, Yury O Chernoff, and Mark Borodovsky. 2008. “Gene Prediction in Novel Fungal Genomes Using an Ab Initio Algorithm with Unsupervised Training.” Genome Research . Cold Spring Harbor Lab, gr--081612.↩
[R16] Hoff, KJ 2019. MakeHub: Fully automated generation of UCSC Genome Browser Assembly Hubs. Genomics, Proteomics and Bioinformatics , in press, preprint on bioarXive, doi: https://doi.org/10.1101/550145.↩
[R17] Bruna, T., Lomsadze, A., & Borodovsky, M. 2020. GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genomics and Bioinformatics, 2(2), lqaa026. doi: https://doi.org/10.1093/nargab/lqaa026.↩
[R18] Kriventseva, EV, Kuznetsov, D., Tegenfeldt, F., Manni, M., Dias, R., Simão, FA, and Zdobnov, EM 2019. OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Research, 47(D1), D807-D811.↩
[R19] Keilwagen, J., Hartung, F., Grau, J. (2019) GeMoMa: Homology-based gene prediction utilizing intron position conservation and RNA-seq data. Methods Mol Biol. 1962:161-177, doi: 10.1007/978-1-4939-9173-0_9.↩
[R20] Keilwagen, J., Wenk, M., Erickson, JL, Schattat, MH, Grau, J., Hartung F. (2016) Using intron position conservation for homology-based gene prediction. Nucleic Acids Research, 44(9):e89.↩
[R21] Keilwagen, J., Hartung, F., Paulini, M., Twardziok, SO, Grau, J. (2018) Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinformatics, 19(1):189.↩
[R22] SRA Toolkit Development Team (2020). SRA Toolkit. https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software.[↩](#a22)
[R23] Kim, D., Paggi, JM, Park, C., Bennett, C., & Salzberg, SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature biotechnology, 37(8):907-915.↩
[R24] Quinlan, AR (2014). BEDTools: the Swiss‐army tool for genome feature analysis. Current protocols in bioinformatics, 47(1):11-12.↩
[R25] Kovaka, S., Zimin, AV, Pertea, GM, Razaghi, R., Salzberg, SL, & Pertea, M. (2019). Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome biology, 20(1):1-13.↩
[R26] Pertea, G., & Pertea, M. (2020). GFF utilities: GffRead and GffCompare. F1000Research, 9.↩
[R27] Huang, N., & Li, H. (2023). compleasm: a faster and more accurate reimplementation of BUSCO. Bioinformatics, 39(10), btad595.↩
[R28] Bruna, T., Gabriel, L. & Hoff, KJ (2024). Navigating Eukaryotic Genome Annotation Pipelines: A Route Map to BRAKER, Galba, and TSEBRA. arXiv, https://doi.org/10.48550/arXiv.2403.19416 .↩


