binnacle
1.0.0
に
Binnacleは、グラフ足場のカバレッジを正確に計算し、MetaBat2、Maxbin 2.0、Concoctなどの主要なビニング方法とシームレスに統合します。ビニングのためのコンティグ(最も一般的なアプローチ)とは対照的に、グラフ足場を使用すると、メタゲノムビンの連続性と品質が向上し、再構築されたゲノムのより広範なセットのセットをキャプチャできます。
Binnacleを実行するには、Python 3.7.x、Bedtools、Samtools、Biopython、NetworkX、Numpy、およびPandasが必要です。
Environment.ymlファイルが利用可能で、これを使用して、Binnacleを実行するのに適したコンドラ環境を作成できます。これらのパッケージをインストールする方法に関する詳細なドキュメントは、こちらをご覧ください。 Metacarvel足場ツールの出力であるグラフ足場を使用するため、Metacarvelをダウンロードしてインストールする必要があります。 Metacarvelのステップバイステップインストールガイドがあります。
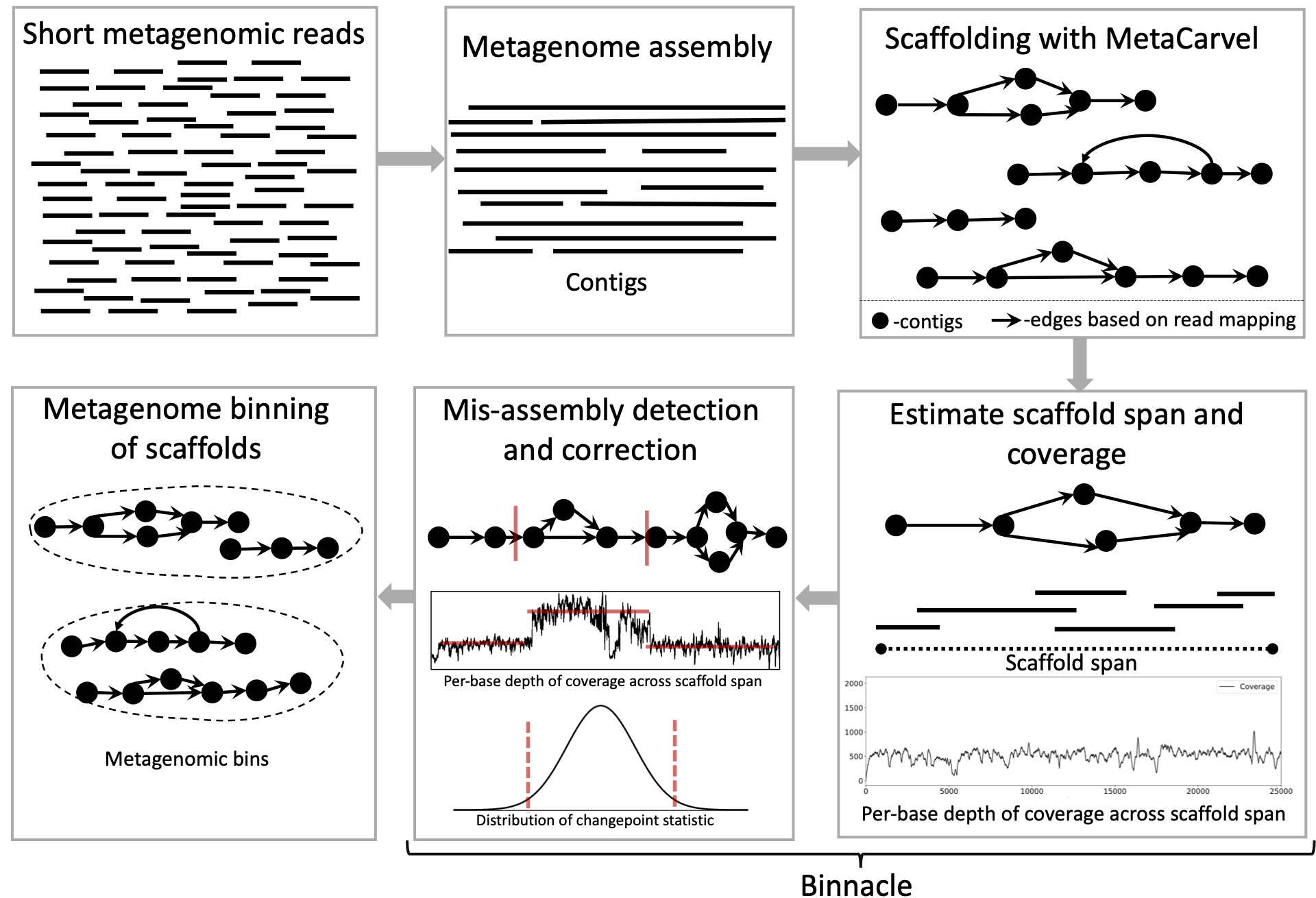
一般に、1つまたは複数のメタゲノムサンプルがある場合は、各サンプルから組み立て、足場、およびビンコンティグ/足場をメタゲノミックビンを生成する必要があります。 Megahitを組み立てに使用し、Metacarvelを足場に使用することをお勧めします。ここでは、アセンブリ、足場、およびベースごとのカバレッジの推定手順を通じて作業するヘルパーガイドを提供します。
これらの手順に従って、グラフ足場でビニング方法を実行するためのファイルを生成します。
python Estimate_Abundances.py -g [ORIENTED.gml] -a [COVERAGE_SORTED.txt] -c [CONTIGS.fa] -d [OUTPUT_DIRECTORY]
usage: Estimate_Abundances.py [-h] [-g ASSEMBLY] [-a COVERAGE] [-bam BAMFILE]
[-bed BEDFILE] [-c CONTIGS] -d DIR [-o COORDS]
[-w WINDOW_SIZE] [-t THRESHOLD]
[-n NEIGHBOR_CUTOFF] [-p POSCUTOFF]
[-pre PREFIX]
binnacle: A tool for binning metagenomic datasets using assembly graphs and
scaffolds generated by metacarvel. Estimate_Abundances.py estimates abundance
for scaffolds generated by MetaCarvel. If the coordinates computed by binnacle
is specified then the abundance for each scaffold is estimated based on the
contig abundances and the coordinates. If the coordinates are not specified
then binnacle etimates the abundance from scratch. While calculating all vs
all abundances please specify the coordinates(Coordinates_After_Delinking.txt)
through the "coords" parameter. The abundances can be provided as a bed file,
bam file or a text file describing the per base coverage obtained by running
the genomeCoverageBed program of the bedtools suite.
optional arguments:
-h, --help show this help message and exit
-g ASSEMBLY, --assembly ASSEMBLY
Assembly Graph generated by Metacarvel
-a COVERAGE, --coverage COVERAGE
Output generated by running genomecov -d on the bed
file generated by MetaCarvel.
-bam BAMFILE, --bamfile BAMFILE
Bam file from aligning reads to contigs
-bed BEDFILE, --bedfile BEDFILE
Bed file from aligning reads to contigs. If bed file
is provided please provide a fasta file of the contigs
-c CONTIGS, --contigs CONTIGS
Contigs generated by the assembler, contigs.fasta
-d DIR, --dir DIR output directory for results
-o COORDS, --coords COORDS
Coordinate file generated by Binnacle
-w WINDOW_SIZE, --window_size WINDOW_SIZE
Size of the sliding window for computing test
statistic to identify changepoints in coverages
(Default=1500)
-t THRESHOLD, --threshold THRESHOLD
Threshold to identify outliers (Default=99)
-n NEIGHBOR_CUTOFF, --neighbor_cutoff NEIGHBOR_CUTOFF
Filter size to identify outliers within (Defualt=100)
-p POSCUTOFF, --poscutoff POSCUTOFF
Position cutoff to consider delinking (Default=100)
-pre PREFIX, --prefix PREFIX
Prefix to be attached to all outputs
-g Path to oriented.gml from running metacarvel on sample
-c Path to contigs obtained by assembling reads of sample
-a Coverage of contigs in ths sample by mapping to its reads -- See Wiki for how to calculate coverage information
-d Output directory
-a Coverage of contigs in Sample 1 by mapping reads of Sample 2 -- See Wiki for how to calculate coverage information
-o Coordinates of scaffolds from Sample 1 that you would have generated from the previous step.
-d Same output directory as Sample 1
python Collate.py -h
usage: Collate.py [-h] -d DIR [-m METHOD] [-k KEEP]
binnacle: A tool for binning metagenomic datasets using assembly graphs and
scaffolds generated by metacarvel.Estimate_Abundances.py estimates abundance
for scaffolds generated by MetaCarvel. The program Collate.py collects the
summary files generated by Estimate_Abundances.py
optional arguments:
-h, --help show this help message and exit
-d DIR, --dir DIR Output directory that contains the summary files
generated by running Estimate_Abundances.py
-m METHOD, --method METHOD
Binning method to format the output to. Presently we
support 1. Metabat 2. Maxbin 3. Concoct 4. Binnacle
(Default)
-k KEEP, --keep KEEP Retain the summary files generated by
Estimate_Abundances.py. Defaults to True
Python環境のセットアップに関する詳細な説明、カバレッジを計算する方法、およびBinnacleを実行する典型的なワークフローについては、Wikiをチェックアウトしてください。
グラフの足場を視覚化するには、WebベースのブラウザであるMetagenomescopeを使用することをお勧めします。 Metagenomescopeへの入力はAssembly_graph_filtered.gmlです。 MetagenomeScopeのインストールと実行に関する詳細なドキュメントは、こちらをご覧ください。
Muralidharan HS、Shah N、Meisel JSおよびPOP M(2021)Binnacle:足場を使用して、メタゲノミックビンの連続性と品質を改善してください。フロント。マイクロビオール。 12:638561。 doi:10.3389/fmicb.2021.638561。
このツールはまだ開発中です。質問がある場合は、Githubでここで問題を開くか、お問い合わせください。
Harihara Muralidharan:[email protected]
Nidhi Shah:[email protected]


