ClockstaR
1.0.0

Sebastian Duchene, Martyna Molak, Simon YW Ho.
분자생태학, 진화 및 계통발생학(MEEP) 연구실
생명과학부
시드니대학교
2015년 6월 10일
partition_data_partitionfinder('drag fasta file with concatenated data here', 'drag partition finder output here')
optim.trees.interactive(folder.parts = 'path to your folder with fasta files and tree topology here')
BSD 거리의 미분을 사용하여 트리 거리 최적화 구현
토폴로지 거리에 대한 병렬 버전 구현
토폴로지 거리 클러스터링에 대한 튜토리얼 작성
모델 테스트를 위한 모델 생성기 통합
분기 길이 및 토폴로지의 최대 가능성 최적화를 위해 RaxML 통합
다중 유전자 데이터 세트를 사용하여 진화 시간 척도를 추정하는 것은 계통발생 연구에서 일반적인 작업입니다. 다중유전자 데이터 세트는 유전자, 코돈 위치 또는 둘 다에 따라 분할될 수 있습니다. 이 튜토리얼에서는 "데이터 하위 집합"을 개별 유전자 또는 다중 유전자 데이터 세트의 하위 단위로 지칭합니다. "파티션"이라는 용어는 데이터 하위 집합 그룹을 나타냅니다.
단일 완화 클럭 모델을 사용하여 데이터 하위 집합을 연결하고 분석할 수 있지만 계보 간 속도 변동 패턴은 트리 토폴로지가 동일한 경우에도 데이터 하위 집합 간에 다를 수 있습니다. 예를 들어, 미토콘드리아 유전자의 계보 간 비율 변화는 핵 유전자의 것과 다를 수 있습니다. 따라서 진화적 시간 척도 및 통계적 적합성의 추정치를 개선하기 위해 다양한 완화 시계 모델을 다양한 데이터 하위 집합에 할당할 수 있습니다(Duchene and Ho., 2014a 참조).
다중 유전자 데이터 세트를 분할할 수 있는 방법에는 여러 가지가 있습니다. 분할 방식을 비교하는 일반적인 접근 방식은 모델 적합성에 대한 베이즈 요인 또는 우도 기반 기준을 사용하는 것입니다. 그러나 대부분의 경우 가능한 모든 분할 방식을 테스트하는 것은 불가능하며, 특히 베이즈 요인을 계산하는 계산 집약적인 방법을 사용하는 경우에는 더욱 그렇습니다.
ClockstaR은 각 데이터 하위 집합의 계통발생 가지 길이를 추정합니다. sBSDmin으로 알려진 분기 점수 거리는 계보 간 비율 변화 패턴의 차이를 측정하기 위해 모든 트리 쌍에 대해 계산됩니다. 이러한 거리는 패키지 클러스터에 구현된 대로 PAM 클러스터링 알고리즘과 함께 GAP 통계를 사용하여 최상의 분할 전략을 추론하는 데 사용됩니다(Maechler et al., 2012)(sBSDmin 메트릭에 대한 자세한 내용은 Duchene et al., 2014b 참조). .
ClockstaR은 다중 유전자 데이터 세트의 계통 발생 분자 시계 분석을 위한 R 패키지입니다. 시계 분할 전략을 선택하기 위해 서로 다른 유전자에 대한 계보 비율 변화 패턴을 사용합니다. 이 방법은 계통발생 트리 거리 측정법과 비지도 기계 학습 알고리즘을 사용하여 최적의 시계 분할 수를 식별하고 각 분할에서 분석해야 하는 유전자를 식별합니다. ClocsktaR에서 선택한 분할 상태는 BEAST, MrBayes, PhyloBayes 등과 같은 프로그램을 사용하여 후속 분자 시계 분석에 사용할 수 있습니다.
원본 출판물을 보려면 이 링크를 따르십시오.
ClockstaR에는 R 설치가 필요합니다. 또한 아래에 설명된 대로 R을 통해 얻을 수 있는 일부 R 종속성이 필요합니다.
요청이나 질문이 있으시면 Sebastian Duchene (sebastian.duchene[at]sydney.edu.au)로 보내주세요. 다른 소프트웨어 및 리소스는 시드니 대학의 분자 생태학, 진화 및 계통 발생학 연구소에서 찾을 수 있습니다.
이 저장소를 zip 파일로 다운로드하고 압축을 풉니다. 다음 지침에서는 일부 fasta 파일과 newick 형식의 계통 발생 트리가 포함된 clockstar_example_data 폴더를 사용합니다. 텍스트 랭글러와 같은 텍스트 편집기에서 이러한 파일을 엽니다. 이 데이터는 진화 속도 변화의 4가지 패턴에 따라 시뮬레이션되었습니다. 트리는 모든 유전자 또는 데이터 파티션에 대한 트리 토폴로지입니다. ClockstaR을 실행하려면 clockstar_example_data의 예제 데이터와 유사한 데이터 형식을 지정하세요.
ClockstaR은 GitHub에서 직접 설치할 수 있습니다. 이를 위해서는 devtools 패키지가 필요합니다. R 프롬프트에 다음 코드를 입력하여 필요한 모든 도구를 설치합니다(패키지를 직접 다운로드하려면 인터넷 연결이 필요합니다).
install . packages ( " devtools " )
library (devtools)
install_github ( ' ClockstaR ' , ' sebastianduchene ' )다운로드 및 설치 후, 함수 라이브러리 와 함께 ClockstaR을 로드합니다.
library (ClockstaR2)프로그램 실행 방법에 대한 예를 보려면 다음을 입력하세요.
example (ClockstaR2)이 튜토리얼의 나머지 부분에서는 clockstar_example_data 폴더를 사용합니다.
첫 번째 단계는 각 정렬에 대한 유전자 나무를 얻는 것입니다. 이를 위해 우리는 트리 토폴로지를 사용하고 각 개별 유전자 정렬(이 경우 A1.fasta부터 C3.fasta까지)을 사용하여 가지 길이를 최적화합니다. 유전자 나무가 있는 경우 파일에 newick 형식으로 저장하고 다음 단계(대화식으로 clockstar 실행)로 이동합니다.
R 프롬프트에 다음 코드를 입력하고 Enter 키를 누릅니다.
optim . trees . interactive ()phangorn 패키지 설치에 대한 오류 메시지가 나타나면 이 코드를 사용한 다음 optim.trees.interactive()를 반복하세요.
install . packcages ( " phangorn " )ClockstaR은 다음 메시지를 인쇄합니다:
Please drag a folder with the data subsets and a tree topology . The files should be in FASTA format, and the trees in NEWICKclockstar_example_data 폴더를 R 콘솔로 드래그하고 Enter를 입력합니다. 폴더에는 FASTA 형식의 알림과 NEWICK의 트리 토폴로지만 포함되어야 합니다. 다음 메시지가 표시됩니다.
What should be the name of the file to save the optimised trees ?최적화된 트리의 파일 이름을 입력합니다. 이 경우 "example.trees"를 사용합니다.
example . trees이 시점에서 ClockstaR은 각 유전자에 대해 별도의 대체 모델을 사용해야 하는지 아니면 모든 경우에 JC를 사용해야 하는지 묻습니다. 이 데이터는 JC에서 시뮬레이션되었으므로 "n"을 입력하고 Enter 키를 누릅니다. 각 대체 모델을 별도로 지정하려면 "y"를 입력합니다.
"n"을 입력하고 Enter를 누르면 ClockstaR이 실행되기 시작합니다. 그래픽 장치에 유전자 나무가 인쇄됩니다. 지정된 트리가 뿌리가 있는 경우에는 무시해도 되는 몇 가지 경고가 인쇄될 수도 있습니다.
clockstar_example_data 폴더를 엽니다. 위의 몇 단계에서 지정한 대로 "example.trees"라는 이름의 파일을 찾을 수 있습니다. 텍스트 편집기에서 example.trees를 엽니다. 여기에는 유전자 정렬의 이름에 따라 각 유전자 나무와 나무 이름이 포함됩니다. 다음과 같아야 합니다.
A1 . fasta (( t1 : 0.01504695462 ,( t2 : 0.00987 ...
A2 . fasta (( t1 : 0.01520523401 ,( t2 : 0.01317 ...
A3 . fasta (( t1 : 0.01519309467 ,( t2 : 0.01092 ...
.
.
.나무가 포함된 이 파일은 다음 단계에서 사용됩니다.
이 단계에서는 이전 단계에서 얻은 것과 같은 파일에 유전자 나무가 있어야 합니다.
R을 열고 위와 같이 ClockstaR을 로드합니다. 프롬프트에 다음 코드를 입력하세요.
clockstar . interactive ()ClockstaR은 다음 메시지를 인쇄합니다:
please drag or type in the path to your gene trees file in NEWICK format :유전자 트리가 포함된 파일을 R 콘솔로 드래그합니다. 이전 단계를 따른 경우 파일 이름은 example.trees입니다. 엔터를 입력하세요.
설치한 패키지에 따라 ClockstaR이 병렬로 실행되어야 하는지 물어볼 수 있습니다. 이는 대규모 데이터 세트에 효율적입니다. 그러나 예제 데이터의 경우 큰 차이가 없으므로 이 메시지가 표시되면 "n"을 입력한 다음 Enter를 입력합니다.
Packages foreach and doParallel are available for parallel computation
Should we run ClockstaR in parallel (y / n) ? (This is good for large data sets)이제 Clockstar가 실행되기 시작합니다. 화면의 출력은 다음과 같아야 합니다.
[ 1 ] " Calculating sBSDmin distances between all pairs of trees "
[ 1 ] " Estimating tree distances "
[ 1 ] " estimating distances 1 of 11 "
[ 1 ] " estimating distances 2 of 11 "
[ 1 ] " estimating distances 3 of 11 "
[ 1 ] " estimating distances 4 of 11 "
[ 1 ] " estimating distances 5 of 11 "
.
.
.원본 출판물에 설명된 나무 거리를 추정한 후 ClockstaR은 다음 메시지를 인쇄합니다.
" I finished calculating the sBSDmin distances between trees "
The settings for clustering with ClockstaR are :
PAM clustering algorithm
K from 1 to number of data subsets - 1
SEmax criterion to select the optimal k
500 bootstrap replicates
Are these correct ? (y / n)클러스터링 알고리즘에 대한 설정입니다. 대부분의 데이터 세트에 적합하므로 이 예에서는 "y"를 입력한 다음 Enter를 입력할 수 있습니다. "n"을 입력하면 이러한 설정을 변경할 수 있습니다. 자세한 내용은 Kaufman 및 Rousseeuw(2009)를 참조하세요.
ClockstaR은 이제 클러스터링 알고리즘을 실행합니다. 마지막에는 최적의 파티션 수를 인쇄하고 결과를 PDF 파일에 저장할지 여부를 묻습니다.
[ 1 ] " ClockstaR has finished running "
[ 1 ] " The best number of partitions for your data set is: 3 "
Do you wish to save the results in a pdf file ? (y / n)"y"를 입력한 후 Enter를 누르세요.
그런 다음 ClockstaR은 출력 파일의 이름을 묻습니다.
What should be the name and path of the output file ?이 예에서는 "example_run"을 입력하고 입력하지만 어떤 이름이라도 사용할 수 있습니다.
이제 clockstar_example_data 폴더를 열고 example_run_gapstats.pdf 및 example_run_matrix.pdf라는 두 개의 PDF 파일을 엽니다.
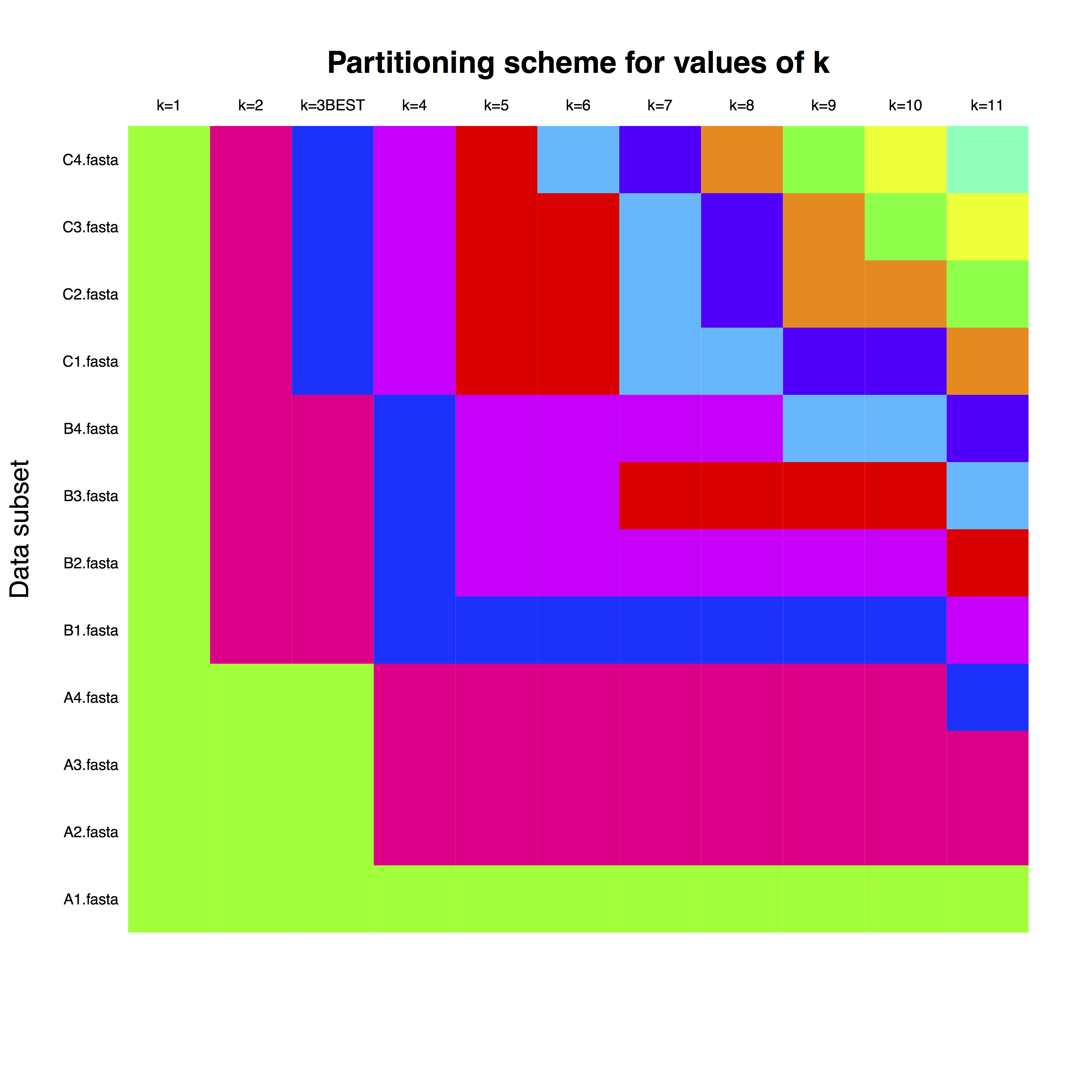
example_run_matrix는 행이 FASTA 파일에 명명된 각 유전자에 해당하는 행렬입니다. 열은 파티션의 수이고, 색상은 시계 파티션에 대한 각 유전자의 할당을 나타냅니다. 예를 들어, 최적의 파티션 수인 k = 3의 경우 문자 A, B 및 C가 있는 유전자에 대해 별도의 시계 파티션을 사용할 수 있습니다.
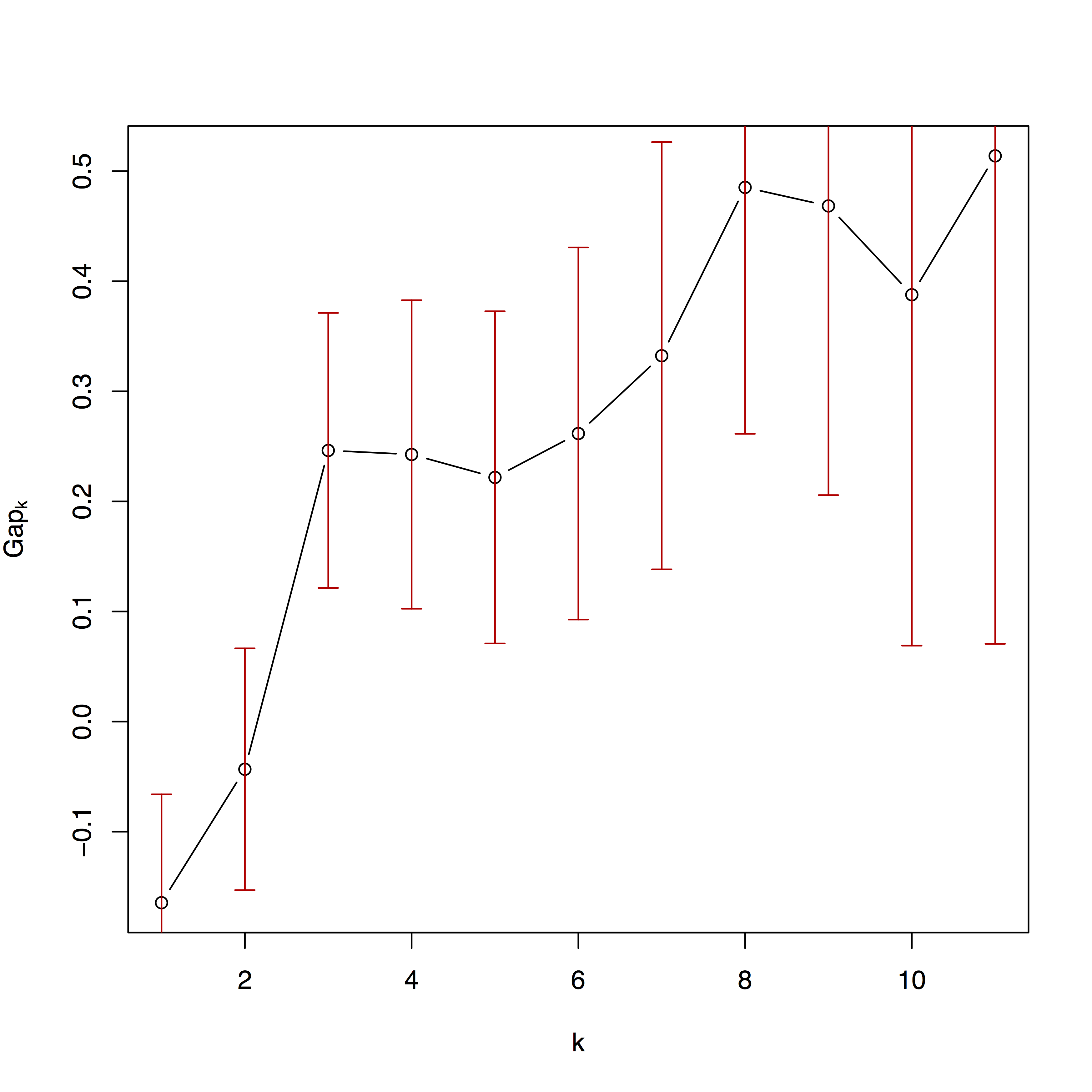
두 번째 플롯은 다양한 수의 파티션에 대한 클러스터링 알고리즘의 적합성입니다. 자세한 내용은 Kaufman 및 Rousseeuw(2009) 및 패키지 클러스터 설명서에서 확인할 수 있습니다.
ClockstaR은 다른 사용자 정의 설정으로 실행할 수 있습니다. 자세한 내용은 문서를 참조하거나 질문이 있는 경우 sebastian.duchene[at]sydney.edy.au로 연락주세요.
로고는 Jun Tong이 디자인했습니다.
Duchene, S., & 호, SY(2014a). 다중 완화 시계 모델을 사용하여 DNA 서열 데이터로부터 진화적 시간 척도를 추정합니다. 분자계통발생학과 진화 (77): 65-70.
Duchene, S., Molak, M., & Ho, SY(2014b). ClockstaR: 분자 계통발생 분석에서 완화된 시계 모델의 수를 선택합니다. 생물정보학 30(7): 1017-1019.
Kaufman, L., & Rousseeuw, PJ (2009). 데이터에서 그룹 찾기: 군집 분석 소개(Vol. 344). 존 와일리 & 선즈.


