reference_database_creator
bug fix --in-silico-pcr --untrimmed
게 ( 기음 반응 아르 자형 참조 데이터베이스 에이 플리콘- 비 애시드 에스 시퀀싱)은 메타게놈 분석을 위해 선별된 참조 데이터베이스를 생성하는 다목적 소프트웨어 프로그램입니다. CRABS 작업흐름은 7개의 모듈로 구성됩니다: (i) 온라인 저장소에서 데이터 다운로드; (ii) 다운로드한 데이터를 CRABS 형식으로 가져옵니다. (iii) 인실리코 PCR 분석을 통해 앰플리콘 영역을 추출하고; (iv) 인실리코 추출 바코드와의 정렬을 통해 프라이머 결합 영역이 없는 앰플리콘을 검색합니다. (v) 여러 필터링 매개변수를 통해 로컬 데이터베이스를 선별하고 부분 집합화합니다. (vi) 분류 분류자 요구 사항에 따라 다양한 형식으로 로컬 데이터베이스를 내보냅니다. (vi) 로컬 참조 데이터베이스의 요약 개요를 탐색하고 제공하기 위한 후처리 기능, 즉 시각화. 이 7개 모듈은 18개 기능으로 나누어져 있으며 아래에 설명되어 있습니다. 또한 18개 함수 각각에 대한 예제 코드가 제공됩니다. 마지막으로 MiFish-E 프라이머 세트에 대한 로컬 상어 참조 데이터베이스를 구축하기 위한 튜토리얼이 이 README 문서의 끝에 제공되어 참조용 예제 스크립트를 제공합니다.
CRABS가 사용자 피드백을 기반으로 대대적인 업데이트와 코드 재설계를 했다는 소식을 발표하게 되어 기쁘게 생각합니다. 이를 통해 자체 로컬 참조 데이터베이스를 구축하는 사용자 경험이 향상되기를 바랍니다!
CRABS v 1.0.0 에 추가된 기능 및 개선 사항 목록은 아래에서 확인하세요.
이제 이 GitHub 저장소를 복제하여 CRABS v 1.0.0을 수동으로 다운로드할 수 있습니다(자세한 정보는 4.1 수동 설치 참조). 최신 버전을 쉽게 설치할 수 있도록 Docker 컨테이너와 conda 패키지를 최대한 빨리 업데이트하겠습니다.
연구 프로젝트에 CRABS를 사용할 때 다음 논문을 인용해 주세요.
[Jeunen, G.-J., Dowle, E., Edgecombe, J., von Ammon, U., Gemmell, N. J., & Cross, H. (2022). crabs—A software program to generate curated reference databases for metabarcoding sequencing data. Molecular Ecology Resources, 00, 1– 14.](https://doi.org/10.1111/1755-0998.13741)
CRABS는 일반적인 Unix/Linux 환경에서 실행되는 명령줄 전용 툴킷이며 Python3으로만 작성되었습니다. 그러나 CRABS는 Python의 하위 프로세스 모듈을 사용하여 bash 구문에서 여러 명령을 실행하여 Python 관련 특이성을 우회하고 실행 속도를 높입니다. CRABS를 설치하는 세 가지 방법을 제공합니다. 최신 버전의 CRABS를 위해서는 이 GitHub 저장소를 복제하고 10개의 종속성을 별도로 설치하여 수동 설치를 권장합니다(모든 종속성에 대한 설치 지침은 4.1 수동 설치에서 제공됨). CRABS는 Docker 및 conda를 통해서도 설치할 수 있습니다. 두 가지 방법 모두 모든 종속 항목을 자동으로 함께 설치하여 쉽게 설치할 수 있습니다. 우리는 Docker 컨테이너와 conda 패키지를 최신 상태로 유지하는 것을 목표로 하고 있지만, 특히 conda 패키지의 경우 최신 버전으로 업데이트하는 데 약간의 지연이 발생할 수 있습니다. 다음은 세 가지 접근 방식 모두에 대한 세부정보입니다.
수동 설치의 경우 먼저 CRABS 저장소를 복제하십시오. 이 단계에서는 명령줄에서 GitHub를 사용할 수 있어야 합니다(GitHub 설치 지침).
git clone https://github.com/gjeunen/reference_database_creator.git
설정에 따라 CRABS를 시스템에서 실행 가능하게 만들어야 할 수도 있습니다. 이는 아래 코드를 사용하여 달성할 수 있습니다.
chmod +x reference_database_creator/crabs
CRABS가 설치되면 모든 종속성이 설치되고 전역적으로 액세스할 수 있는지 확인해야 합니다. CRABS의 최신 버전(버전 v 1.0.0 )은 Python 3.11.7(또는 3.11.7과 호환되는 모든 버전)에서 실행되며 Python에 표준으로 제공되지 않을 수 있는 5개의 Python 모듈과 5개의 외부 소프트웨어 프로그램을 사용합니다. 모든 종속성은 설치 지침에 대한 링크와 함께 아래에 나열되어 있습니다. 각 모듈 및 소프트웨어 프로그램에 제공되는 버전 번호는 CRABS가 개발된 버전 번호입니다. 각각의 호환 가능한 버전도 사용할 수 있습니다.
Python 모듈:
외부 소프트웨어 프로그램:
CRABS와 모든 종속 항목이 설치되면 아래 코드를 사용하여 OS 전체에서 CRABS에 액세스할 수 있습니다.
export PATH="/path/to/crabs/folder:$PATH"
/path/to/crabs/folder를 OS의 GitHub 저장소 폴더에 대한 실제 경로, 즉 위의 git clone 명령 중에 생성된 폴더로 대체하세요. .bash_profile 또는 .bashrc 파일 에 export 코드를 추가하면 언제든지 CRABS에 전역적으로 액세스할 수 있습니다.
Docker는 컴퓨터에서 격리되어 Docker 엔진이라는 가상 호스트 운영 체제를 통해 실행되는 '컨테이너' 내에 소프트웨어 애플리케이션을 배포할 수 있는 오픈 소스 프로젝트입니다. 가상 머신에 비해 docker를 실행하는 주요 이점은 훨씬 적은 리소스를 사용한다는 것입니다. 이러한 격리는 Mac, Windows, Linux를 포함한 대부분의 운영 체제에서 Docker 컨테이너를 실행할 수 있음을 의미합니다. Docker Desktop을 사용하려면 무료 계정을 설정해야 할 수도 있습니다. 이 링크에는 Docker 사용의 기본 사항이 잘 소개되어 있습니다. 다음은 Docker 멀티버스를 시작하고 방향을 잡을 수 있는 링크입니다.
컴퓨터에서 Crabs를 실행하려면 두 단계만 거치면 됩니다. 먼저 대부분의 사용자에게 무료로 제공되는 Docker Desktop을 컴퓨터에 설치합니다. Mac에 대한 지침은 다음과 같습니다 . 다음은 Windows 컴퓨터에 대한 지침이고 , Linux에 대한 지침은 다음과 같습니다 (대부분의 주요 Linux 플랫폼이 지원됩니다). Docker Desktop을 설치하고 실행한 후(명령줄에서 docker 명령을 사용하려면 데스크톱 애플리케이션이 실행 중이어야 함) Crabs 이미지를 '풀'하기만 하면 바로 사용할 수 있습니다.
docker pull quay.io/swordfish/crabs:0.1.7
Docker 애플리케이션 설치는 쉽지만 해당 애플리케이션을 사용하는 것은 처음에는 약간 까다로울 수 있습니다. 시작하는 데 도움이 되도록 Docker 버전의 crabs를 사용하여 몇 가지 예제 명령을 제공했습니다. 이러한 예제는 이 저장소의 docker_intro 폴더에서 찾을 수 있습니다 . 이러한 예를 통해 전체 참조 데이터베이스 설정을 실행하고 준비를 완료할 수 있습니다. 우리는 이러한 예제를 계속해서 확장하고 다양한 상황에서 이를 테스트할 것입니다. 문제 탭에서 질문하고 피드백을 제공하세요.
conda 패키지를 설치하려면 먼저 conda를 설치해야 합니다. 자세한 내용은 이 링크를 참조하세요. conda가 이미 설치되어 있는 경우 CRABS를 설치하기 전에 conda update conda 로 conda 도구를 업데이트하는 것이 좋습니다.
conda가 설치되면 아래 단계에 따라 CRABS 및 모든 종속성을 설치하십시오. 아래에 나타나는 순서대로 명령을 입력하십시오.
conda create -n CRABS
conda activate CRABS
conda config --add channels bioconda
conda config --add channels conda-forge
conda install -c bioconda crabs
설치 명령을 입력하면 conda는 요청을 처리한 다음(1분 정도 걸릴 수 있음) 설치될 모든 패키지와 프로그램을 표시하고 이를 확인하도록 요청합니다. y 입력하여 설치를 시작합니다. 이 작업이 끝나면 CRABS를 사용할 준비가 되어 있어야 합니다.
우리는 Mac 및 Linux 시스템에서 이 설치를 테스트했습니다. 아직 WSL(Linux용 Windows 하위 시스템)에서는 테스트하지 않았습니다.
아래 코드를 사용하여 CRABS가 성공적으로 설치되었는지 확인하고 도움말 정보를 확인하세요.
crabs -h도움말 정보는 18개의 기능을 여러 그룹으로 나누어 각 그룹의 맨 위에는 기능이 나열되고 그 아래에는 필수 및 선택적 매개변수가 나열됩니다.

CRABS에는 18개의 기능을 통합하는 7개의 모듈이 포함되어 있습니다.
모듈 1: 온라인 저장소에서 데이터 다운로드
--download-taxonomy : NCBI 분류 정보를 다운로드합니다.--download-bold : 생명 바코드 데이터베이스에서 시퀀스 데이터를 다운로드합니다(굵은 글씨);--download-embl : European Nucleotide Archive(ENA, EMBL)에서 서열 데이터를 다운로드합니다.--download-mitofish : MitoFish 데이터베이스에서 시퀀스 데이터를 다운로드합니다.--download-ncbi : NCBI(National Center for Biotechnology Information)에서 시퀀스 데이터를 다운로드합니다.모듈 2: 다운로드한 데이터를 CRABS 형식으로 가져오기
--import : 다운로드한 시퀀스 또는 사용자 정의 바코드를 CRABS 형식으로 가져옵니다.--merge : 다양한 CRABS 형식의 파일을 단일 파일로 병합합니다.모듈 3: in silico PCR 분석을 통해 앰플리콘 영역 추출
--in-silico-pcr : 프라이머 결합 영역을 찾아 제거하여 다운로드한 데이터에서 앰플리콘을 추출합니다.모듈 4: 프라이머 결합 영역 없이 앰플리콘 검색
--pairwise-global-alignment : 다운로드한 시퀀스를 in silico 추출 바코드에 정렬하여 프라이머 결합 영역이 없는 앰플리콘을 검색합니다.모듈 5: 여러 필터링 매개변수를 통해 로컬 데이터베이스 선별 및 하위 집합
--dereplicate : 중복된 시퀀스를 삭제합니다.--filter : 여러 필터링 매개변수를 통해 시퀀스를 삭제합니다.--subset : 지정된 분류 그룹을 유지하거나 제외하기 위해 로컬 데이터베이스의 하위 집합을 설정합니다.모듈 6: 로컬 데이터베이스 내보내기
--export : 사용할 분류 분류기의 요구 사항에 따라 CRABS 형식의 데이터베이스를 다양한 형식으로 내보냅니다.모듈 7: 로컬 참조 데이터베이스의 요약 개요를 탐색하고 제공하는 후처리 기능
--diversity-figure : 참조 데이터베이스에 포함된 지정된 수준당 종 및 서열 그룹의 수를 표시하는 수평 막대 차트를 생성합니다.--amplicon-length-figure : 분류학적 그룹으로 구분된 앰플리콘 길이 분포를 묘사하는 선형 차트를 생성합니다.--phylogenetic-tree : 대상 종 목록에 대한 참조 데이터베이스의 바코드를 사용하여 계통 발생 트리를 생성합니다.--amplification-efficiency-figure : 프라이머 결합 영역의 불일치를 표시하는 막대 그래프를 생성합니다.--completeness-table : 분류 그룹에 대한 바코드 가용성이 포함된 스프레드시트를 생성합니다.초기 시퀀싱 데이터는 (i) 굵게, (ii) EMBL, (iii) MitoFish 및 NCBI를 포함한 4개의 온라인 저장소에서 CRABS를 통해 다운로드할 수 있습니다. 버전 v 1.0.0 부터 각 저장소의 데이터 다운로드는 자체 기능으로 분할됩니다. 또한 CRABS는 유연성을 높이고 데이터 다운로드 실패 시 디버깅을 활성화하기 위해 다운로드 후 데이터 형식을 자동으로 지정하지 않습니다.
CRABS는 서열 데이터를 다운로드하는 것 외에도 CRABS가 각 서열에 대한 분류 계보를 생성하는 데 사용하는 NCBI 분류 정보를 다운로드할 수도 있습니다.
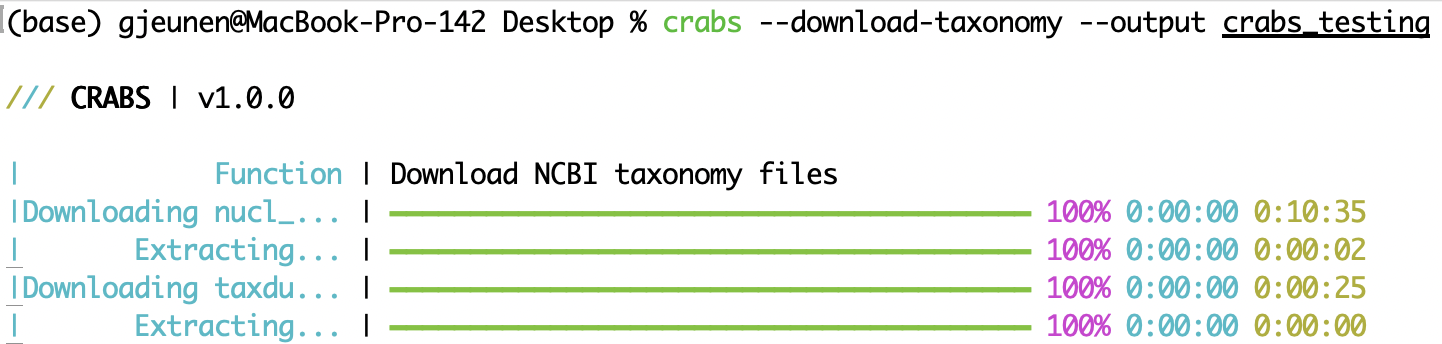
--download-taxonomy 참조 데이터베이스(5.2 모듈 2 참조)의 다운로드된 각 서열에 분류학적 계보를 할당하려면 분류학적 정보를 다운로드해야 합니다. CRABS는 NCBI의 분류법을 활용하고 세 가지 특정 파일을 컴퓨터에 다운로드합니다. (i) 분류 ID에 접근 번호를 연결하는 파일( nucl_gb.accession2taxid ), (ii) 각 분류 ID와 연관된 계통발생 이름에 대한 정보가 포함된 파일( names.dmp ) 및 (iii) 분류학적 ID가 연결되는 방법에 대한 정보가 포함된 파일( nodes.dmp )입니다. 다운로드된 파일의 출력 디렉터리는 --output 매개변수를 사용하여 지정할 수 있습니다. nucl_gb.accession2taxid 파일이나 names.dmp 및 node.dmp 파일을 제외하려면 --exclude acc2tax 또는 --exclude taxdump 매개변수를 각각 제공할 수 있습니다. --exclude 매개변수에 acc2tax 및 taxdump 모두 제공되므로 아래 첫 번째 코드는 파일을 다운로드하지 않습니다. 코드의 두 번째 줄은 세 파일을 모두 --output crabs_testing 하위 디렉터리에 다운로드합니다. 아래 스크린샷은 이 코드 줄을 실행할 때 콘솔에 인쇄되는 내용을 보여줍니다.
crabs --download-taxonomy --exclude 'acc2taxid,taxdump'
crabs --download-taxonomy --output crabs_testing
--download-bold 굵게 표시된 시퀀스는 굵게 표시된 웹사이트를 통해 다운로드됩니다. 두 줄의 fasta 문서로 구성된 출력 파일은 --output 매개변수를 사용하여 지정할 수 있습니다. 사용자는 --taxon 매개변수를 사용하여 다운로드할 분류 그룹을 지정할 수 있습니다. 사용자가 여러 분류 그룹을 다운로드하려는 경우 간단한 for 루프(아래 제공된 예)를 작성하여 인스턴스당 굵게에서 다운로드할 데이터 양을 제한하는 것이 좋습니다. 그러나 제한된 수의 분류 그룹만 관심 대상인 경우 분류 그룹 이름은 | 로 구분할 수도 있습니다. (아래에 제공된 예). 또한 다운로드할 분류 그룹 이름이 굵게 표시된 아카이브에 나열되어 있는지 또는 대체 이름을 사용해야 하는지 사용자에게 확인하는 것이 좋습니다. 예를 들어 --taxon Chondrichthyes 지정하면 이 클래스 이름이 굵게 표시되지 않으므로 굵게 표시된 모든 연골 어류 시퀀스를 다운로드하지 않습니다. 이 경우 사용자는 --taxon Elasmobranchii 사용해야 합니다. 사용자는 --marker 매개변수를 제공하여 특정 유전자 마커에 대한 다운로드를 제한하도록 지정할 수도 있습니다. 여러 유전자 마커에 관심이 있는 경우 마커 이름을 | 로 구분해야 합니다. . 굵게 표시되는 네 가지 주요 DNA 바코딩 마커는 COI-5P , ITS , matK 및 rbcL 입니다. --marker 매개변수에 대한 입력은 대소문자를 구분합니다.
권장 접근 방식: 여러 분류 그룹에 대해 굵게에서 데이터를 다운로드하는 간단한 for 루프(권장 접근 방식). 아래 코드는 먼저 Elasmobranchii에 대한 데이터를 다운로드한 다음 Mammalia에 할당된 시퀀스를 다운로드합니다. 다운로드된 데이터는 --output crabs_testing 하위 디렉토리에 기록되고 두 개의 별도 파일에 배치되어 어떤 데이터가 어떤 분류 그룹에 속하는지 나타냅니다(예: crabs_testing/bold_Elasmobranchii.fasta 및 crabs_testing/bold_Mammalia.fasta .
for taxon in Elasmobranchii Mammalia; do crabs --download-bold --taxon ${taxon} --output crabs_testing/bold_${taxon}.fasta; done

대체 옵션: 권장되는 for 루프 외에도 | 사용하여 이름을 구분하여 여러 분류 이름을 한 번에 제공할 수 있습니다. .
crabs --download-bold --taxon 'Elasmobranchii|Mammalia' --output crabs_testing/bold_elasmobranchii_mammalia.fasta
--download-embl EMBL의 시퀀스는 ENA FTP 사이트를 통해 다운로드됩니다. EMBL 파일은 먼저 '.fasta.gz' 형식으로 다운로드되며 다운로드가 완료되면 자동으로 압축이 풀립니다. 이 데이터베이스는 굵게 또는 NCBI에 비해 선택적 다운로드와 관련하여 많은 유연성을 제공하지 않습니다. 오히려 EMBL 데이터는 15개의 세금 부문으로 구성되어 있으며 별도로 다운로드할 수 있습니다. 다운로드할 세금 구분은 --taxon 매개변수를 사용하여 지정할 수 있습니다. 각 과세 구분이 여러 개의 파일로 분할되어 있으므로 이름 뒤에 * 를 붙여 모든 파일을 다운로드할 수 있습니다. 사용자는 파일 이름 전체를 작성하여 특정 파일을 다운로드할 수도 있습니다. 15가지 세금 분할 옵션 전체 목록이 아래에 나와 있습니다. --output 매개변수를 사용하여 출력 디렉터리와 파일 이름을 지정할 수 있습니다.
세금 구분 목록:
crabs --download-embl --taxon 'mam*' --output crabs_testing/embl_mam.fasta
--download-mitofish CRABS는 MitoFish 데이터베이스를 다운로드할 수도 있습니다. 이 데이터베이스는 두 줄로 구성된 단일 fasta 파일입니다. --output 매개변수를 사용하여 출력 디렉터리와 파일 이름을 지정할 수 있습니다.
crabs --download-mitofish --output crabs_testing/mitofish.fasta

--download-ncbi NCBI 데이터베이스의 시퀀스는 Entrez 프로그래밍 유틸리티를 통해 다운로드됩니다. NCBI를 사용하면 사용자가 --database 매개변수를 사용하여 지정할 수 있는 다양한 데이터베이스에서 데이터를 다운로드할 수 있습니다. 대부분의 사용자에게 --database nucleotide 데이터베이스는 로컬 참조 데이터베이스를 구축하는 데 가장 적합합니다.
NCBI에서 다운로드할 데이터를 지정하기 위해 사용자는 --query 매개변수를 통해 검색을 제공합니다. 좋은 NCBI 검색을 작성하는 것은 어려울 수 있습니다. 검색어를 작성하는 좋은 방법은 NCBI 웹페이지 검색창을 사용하는 것입니다. 이 링크에서 먼저 초기 검색을 수행하고 Enter를 누르십시오. 그러면 검색을 더욱 세분화할 수 있는 결과 페이지가 나타납니다. 아래 스크린샷에서는 서열 길이를 100~25,000bp 사이로 제한하고 미토콘드리아 서열만 통합하여 검색을 더욱 구체화했습니다. 사용자는 웹사이트의 "세부 정보 검색" 상자에 텍스트를 복사하여 붙여넣고 --query 매개변수에 따옴표를 묶어 제공할 수 있습니다. NCBI 웹페이지 검색 창을 사용하는 또 다른 이점은 웹페이지가 검색어와 일치하는 시퀀스 수를 표시한다는 것입니다. 이는 CRABS에서 보고한 시퀀스 수와 일치해야 합니다. 이 웹페이지는 우리 팀이 추가 정보를 위해 작성한 NCBI 웹페이지의 검색 기능 사용에 대한 간단한 추가 튜토리얼을 제공합니다.
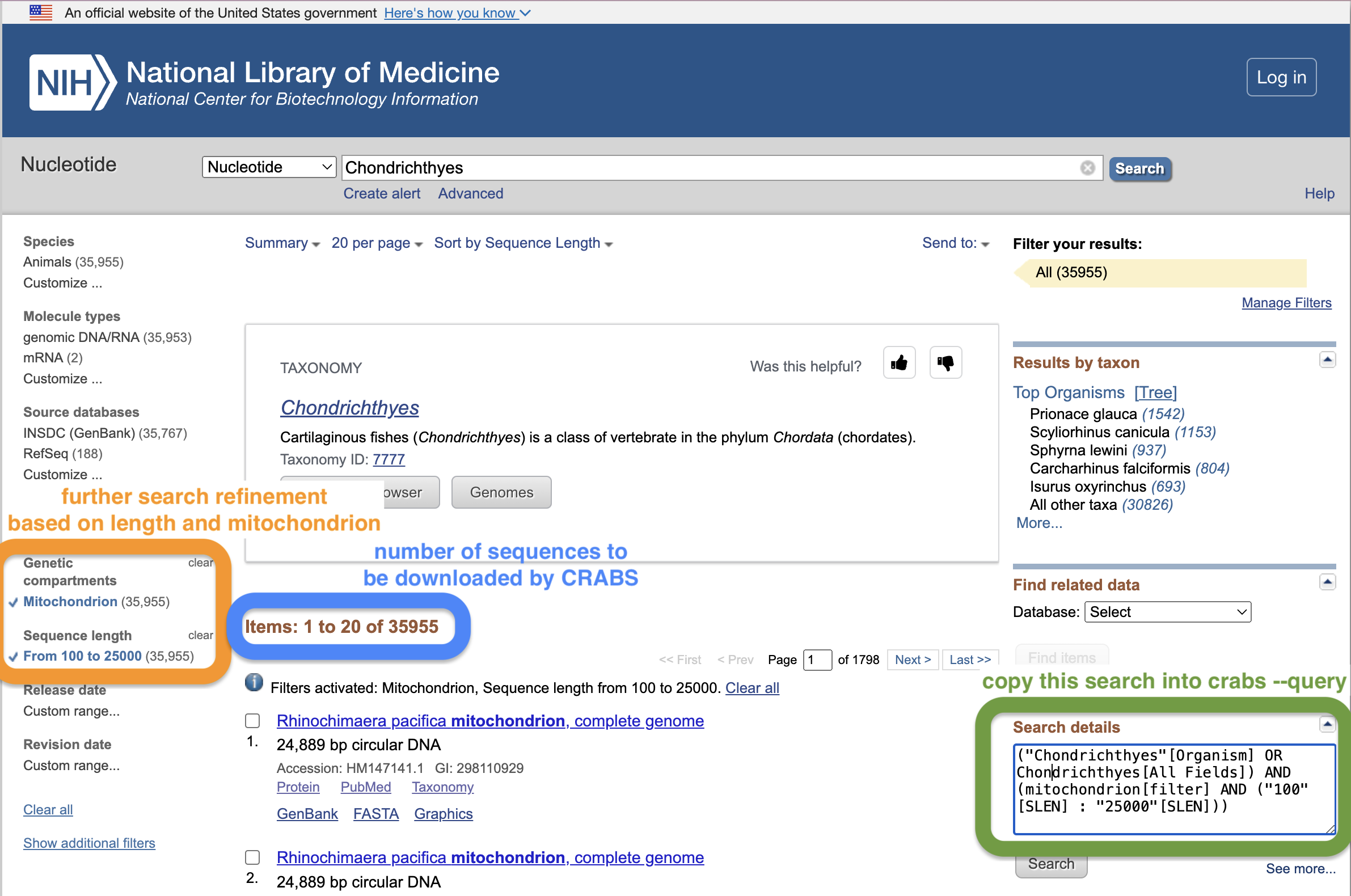
검색 쿼리( --query ) 외에도 사용자는 --species 매개변수를 사용하여 종 목록에 대한 서열 데이터를 다운로드하여 검색어를 추가로 제한할 수 있습니다. --species 매개변수는 + 로 구분된 종 이름의 입력 문자열 또는 문서의 한 줄에 단일 종 이름이 포함된 입력 .txt 파일을 사용합니다. --batchsize 매개변수는 사용자에게 NCBI 웹 사이트에서 N 배치로 시퀀스를 다운로드할 수 있는 옵션을 제공합니다. 이 매개변수의 기본값은 5,000입니다. 한 번에 너무 많은 시퀀스를 다운로드하면 NCBI 서버에서 다운로드 연결을 끊을 가능성이 높으므로 이 값을 5,000 이상으로 늘리는 것은 권장되지 않습니다. --email 매개변수를 사용하면 사용자가 NCBI 서버에 액세스하는 데 필요한 이메일 주소를 지정할 수 있습니다. 마지막으로 --output 매개변수를 사용하여 출력 디렉터리와 파일 이름을 지정할 수 있습니다.
crabs --download-ncbi --query '("Chondrichthyes"[Organism] OR Chondrichthyes[All Fields]) AND (mitochondrion[filter] AND ("100"[SLEN] : "25000"[SLEN]))' --output crabs_testing/ncbi_chondrichthyes.fasta --email [email protected] --database nucleotide

--import 온라인 저장소의 데이터가 다운로드되면 --import 기능을 사용하여 파일을 CRABS로 가져와야 합니다. CRABS 형식은 (i) 시퀀스 ID, (ii) 초기 다운로드에서 구문 분석된 분류 이름, (iii) NCBI 분류 ID 번호, (iv) NCBI 분류에 따른 분류 계보를 포함한 모든 정보를 포함하는 시퀀스당 단일 탭으로 구분된 줄로 구성됩니다. , 및 (v) 순서. CRABS는 각 시퀀스에 대한 NCBI 접근 번호를 시퀀스 ID로 얻으려고 시도합니다. 서열에 접근 번호가 포함되어 있지 않은 경우, 즉 NCBI에 기탁되지 않은 경우 CRABS는 crabs_*[num]*_taxonomic_name 형식을 사용하여 고유한 서열 ID를 생성합니다. 입력 문서의 형식은 --import-format 매개변수를 사용하여 지정되며 데이터가 다운로드된 저장소의 이름(예: BOLD , EMBL , MITOFISH 또는 NCBI ) 을 지정합니다. CRABS가 생성하는 분류 계보는 NCBI 분류를 기반으로 하며 CRABS에는 --download-taxonomy 기능을 사용하여 다운로드한 세 개의 파일(예: --names , --nodes 및 --acc2tax 이 필요합니다. 버전 v 1.0.0 부터 CRABS는 동의어와 허용되지 않는 이름을 확인하여 로컬 참조 데이터베이스에 더 많은 수의 시퀀스와 다양성을 통합할 수 있습니다. 분류 계보에 포함될 분류 순위는 --ranks 매개변수를 사용하여 지정할 수 있습니다. 모든 분류학적 순위를 포함할 수 있지만 대부분의 분류학적 분류자 --ranks 'superkingdom;phylum;class;order;family;genus;species' 에 필요한 모든 정보를 포함하려면 다음 입력을 사용하는 것이 좋습니다. 출력 파일은 --output 매개변수를 사용하여 지정할 수 있으며 간단한 .txt 파일입니다. 터미널 창에서 CRABS는 가져온 시퀀스 수의 결과와 분류학적 계보를 생성할 수 없는 모든 시퀀스를 인쇄합니다.
crabs --import --import-format bold --input crabs_testing/bold_Elasmobranchii.fasta --names crabs_testing/names.dmp --nodes crabs_testing/nodes.dmp --acc2tax crabs_testing/nucl_gb.accession2taxid --output crabs_testing/crabs_bold.txt --ranks 'superkingdom;phylum;class;order;family;genus;species'

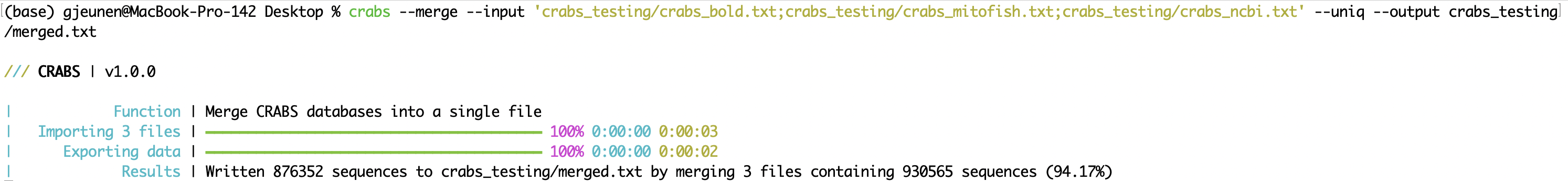
--merge 여러 온라인 저장소의 시퀀스 데이터를 다운로드하는 경우 --merge 기능을 사용하여 파일을 가져온 후(5.2.1 --import 참조) 단일 파일로 병합할 수 있습니다. 병합할 입력 파일은 --input 매개변수를 사용하여 입력할 수 있으며, 파일은 ; 로 구분됩니다. . 다양한 온라인 저장소에 저장될 때 시퀀스가 여러 번 다운로드되었을 가능성이 있습니다. --uniq 매개변수를 사용하면 각 접근 번호의 단일 버전만 유지됩니다. --output 매개변수를 사용하여 출력 파일을 지정할 수 있습니다. 터미널 창에서 CRABS는 병합된 시퀀스 수의 결과와 --uniq 매개변수를 사용할 때 유지되는 시퀀스 수를 인쇄합니다.
crabs --merge --input 'crabs_testing/crabs_bold.txt;crabs_testing/crabs_mitofish.txt;crabs_testing/crabs_ncbi.txt' --uniq --output crabs_testing/merged.txt
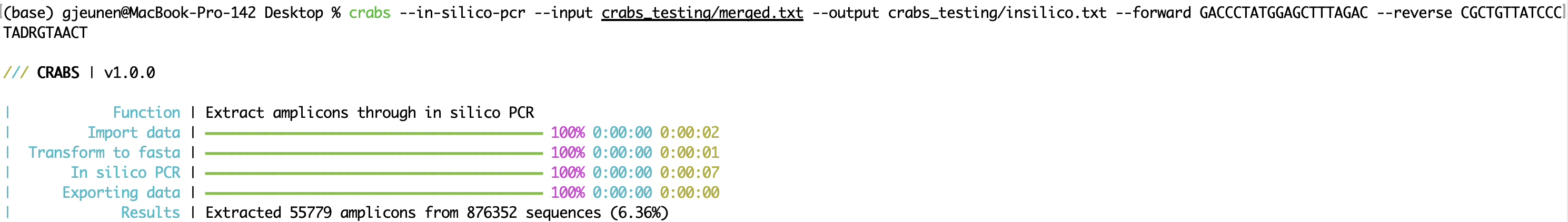
CRABS는 in silico PCR(기능: --in-silico-pcr )을 수행하여 프라이머 세트의 앰플리콘 영역을 추출합니다. CRABS는 in silico PCR에 cutadapt v 4.4를 사용하여 기존 Python 코드의 실행 속도를 높입니다. 입력 및 출력 파일 이름은 각각 ' --input ' 및 ' --output ' 매개변수를 사용하여 지정할 수 있습니다. 정방향 및 역방향 프라이머는 각각 ' --forward ' 및 ' --reverse ' 매개변수를 사용하여 5'-3' 방향으로 제공되어야 합니다. CRABS는 역방향 프라이머를 역보완합니다. 버전 v 1.0.0 부터 CRABS는 단일 in silico PCR 분석을 사용하여 양방향으로 바코드를 유지할 수 있습니다. 따라서 역보완 단계와 in silico PCR의 재실행이 수행되지 않아 실행 속도가 크게 향상됩니다. 프라이머 결합 영역을 찾을 수 없는 시퀀스를 유지하려면 --untrimmed 매개변수에 출력 파일을 지정할 수 있습니다. 프라이머 결합 영역에서 발견된 최대 허용 불일치 수는 --mismatch 매개변수를 사용하여 지정할 수 있으며 기본 설정은 4입니다. 마지막으로 in silico PCR 분석은 CRABS에서 다중 스레드될 수 있습니다. 기본적으로 최대 스레드 수가 사용되지만 사용자는 --threads 매개변수와 함께 사용할 스레드 수를 지정할 수 있습니다.
crabs --in-silico-pcr --input crabs_testing/merged.txt --output crabs_testing/insilico.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT
온라인 데이터베이스에 보관할 때 참조 서열에서 프라이머 결합 영역을 제거하는 것이 일반적인 관행입니다. 따라서 참조 서열이 --in-silico-pcr 함수에서 검색된 것과 동일한 정방향 및/또는 역방향 프라이머를 사용하여 생성된 경우 --in-silico-pcr 기능은 의 앰플리콘 영역을 복구하는 데 실패하게 됩니다. 참조 시퀀스. 이러한 가능성을 설명하기 위해 CRABS에는 VSEARCH v 2.16.0을 사용하여 구현된 쌍별 글로벌 정렬(Pairwise Global Alignment)을 실행하여 참조 서열에 전체 정방향 및 역방향 프라이머 결합 영역이 포함되지 않은 앰플리콘 영역을 추출하는 옵션이 있습니다. 이를 달성하기 위해 --pairwise-global-alignment 함수는 --input 매개변수를 사용하여 원래 다운로드된 데이터베이스 파일을 가져옵니다. 검색할 데이터베이스는 --in-silico-pcr 의 출력 파일이며 --amplicons 매개변수를 사용하여 지정할 수 있습니다. --output 매개변수를 사용하여 출력 파일을 지정할 수 있습니다. 염기쌍 길이를 계산하는 데에만 사용되는 프라이머 서열은 --forward 및 --reverse 매개변수를 사용하여 설정할 수 있습니다. --pairwise-global-alignment 기능은 대규모 데이터베이스에서 실행하는 데 오랜 시간이 걸릴 수 있으므로 --size-select 매개변수를 사용하여 프로세스 속도를 높이기 위해 시퀀스 길이를 제한할 수 있습니다. --percent-identity 및 --coverage 매개변수를 각각 사용하여 최소 백분율 ID 및 쿼리 적용 범위를 지정할 수 있습니다. --percent-identity 0과 1 사이의 백분율 값(예: 95% = 0.95)으로 제공되어야 하며, --coverage 0과 100 사이의 백분율 값(예: 95% = 95)으로 제공되어야 합니다. 기본적으로 --pairwise-global-alignment 기능은 프라이머 서열이 참조 서열에 완전히 존재하지 않는 서열(정방향 또는 역방향 프라이머의 길이 내에서 시작하거나 끝나는 정렬)을 유지하도록 제한됩니다. --all-start-positions 매개변수가 제공되면 정렬이 프라이머 결합 영역 범위 외부에서 발견되면 포지티브 히트가 포함됩니다(너무 많은 불일치로 인해 --in-silico-pcr 함수에서 누락됨). 프라이머 결합 영역). 프라이머에 4개 이상의 불일치가 있는 경우 --in-silico-pcr 함수의 지정된 프라이머 세트를 사용하여 바코드가 증폭될 가능성이 거의 없으므로 --all-start-positions 사용을 권장하지 않습니다. 바인딩 영역.
crabs --pairwise-global-alignment --input crabs_testing/merged.txt --amplicons crabs_testing/insilico.txt --output crabs_testing/aligned.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT --size-select 10000 --percent-identity 0.95 --coverage 0.95
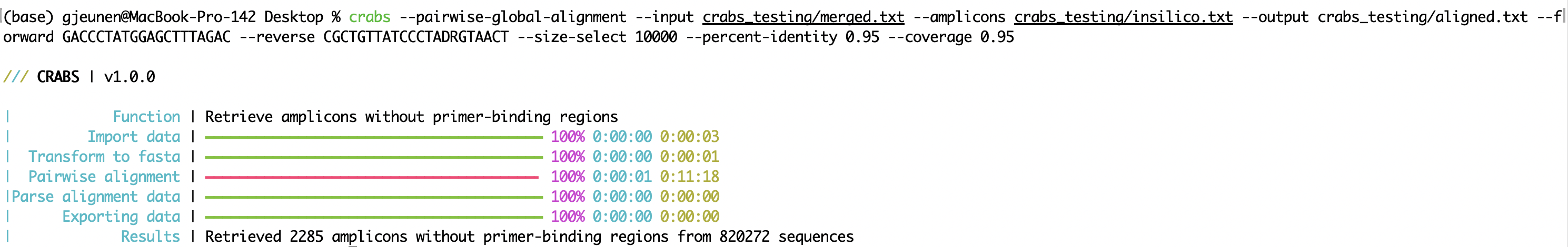
--pairwise-global-alignment 에 대한 코드 실행 속도 향상 --pairwise-global-alignment 함수는 멀티스레딩이 지원되더라도 CRABS가 대규모 시퀀스 파일을 처리할 때 실행하는 데 상당한 시간이 걸릴 수 있습니다. CRABS v 1.0.0 업데이트 이후 --import 에서 --export 까지 동일한 파일 구조가 적용되어 어떤 순서로든 기능을 실행할 수 있습니다. CRABS 워크플로의 순서를 따르는 것이 여전히 권장되지만, --in-silico-pcr 함수 이전에 --dereplicate 및 --filter 함수를 실행하면 --pairwise-global-alignment 함수의 속도가 크게 향상될 수 있습니다. --in-silico-pcr 이전에 이러한 큐레이션 단계를 실행하면 --pairwise-global-alignment 기능을 위해 CRABS에서 처리하는 데 필요한 시퀀스 수가 크게 줄어듭니다.
참고 1 : --in-silico-pcr 이전에 --filter 기능을 실행하는 경우, 시퀀스에 직접 영향을 미치는 매개변수를 생략하십시오. --filter 추출된 앰플리콘이 아닌 전체 시퀀스를 기반으로 하기 때문입니다. . 따라서 --minimum-length , --maximum-length , --maximum-n 매개변수는 생략하세요.
참고 2 : --in-silico-pcr 이전에 --dereplicate 및 --filter 기능을 실행하는 경우 --pairwise-global-alignment 이후에 두 기능을 모두 다시 실행하는 것이 좋습니다. 이제 데이터베이스를 추가로 관리할 수 있기 때문입니다. 앰플리콘이 추출된다는 것입니다.
--in-silico-pcr 및 --pairwise-global-alignment 기능을 통해 프라이머 세트에 대한 모든 잠재적 바코드가 추출되면 로컬 참조 데이터베이스는 --dereplicate 포함한 다양한 기능을 사용하여 CRABS 내에서 추가 큐레이션 및 하위 설정을 거칠 수 있습니다. , --filter 및 --subset .
--dereplicate 첫 번째 큐레이션 방법은 --dereplicate 함수를 사용하여 로컬 참조 데이터베이스를 복제 해제하는 것입니다. 특정 분류군에 대해 이 시점에서 로컬 참조 데이터베이스 내에 여러 개의 동일한 바코드가 포함될 수 있습니다. 이는 서로 다른 연구 그룹이 동일한 서열을 기탁했거나 분류군에 대한 서열 간의 특정 내부 변이가 추출된 바코드에 포함되지 않은 경우 발생할 수 있습니다. 분류 할당 속도를 높이고 분류 할당 결과를 개선하려면 이러한 동일한 참조 바코드를 제거하는 것이 가장 좋습니다(특히 제한된 수의 결과를 제공하는 분류 분류자(예: BLAST)의 경우).
입력 및 출력 파일은 각각 --input 및 --output 매개변수를 사용하여 지정할 수 있습니다. CRABS는 다음을 포함하여 --dereplication-method 매개변수를 사용하여 지정할 수 있는 세 가지 복제 해제 방법을 제공합니다.
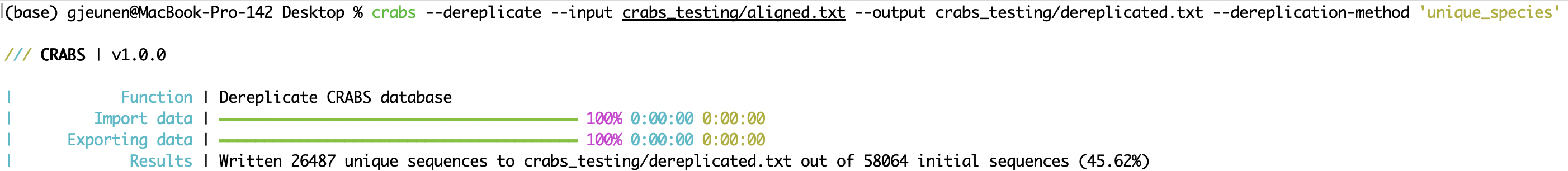
crabs --dereplicate --input crabs_testing/aligned.txt --output crabs_testing/dereplicated.txt --dereplication-method 'unique_species'
--filter 두 번째 큐레이션 방법은 --filter 함수를 사용하여 다양한 매개변수를 사용하여 로컬 참조 데이터베이스를 필터링하는 것입니다. 입력 및 출력 파일은 각각 --input 및 --output 매개변수를 사용하여 지정할 수 있습니다. 버전 v 1.0.0 부터. CRABS는 다음을 포함한 6개 매개변수를 기반으로 필터링을 통합합니다.
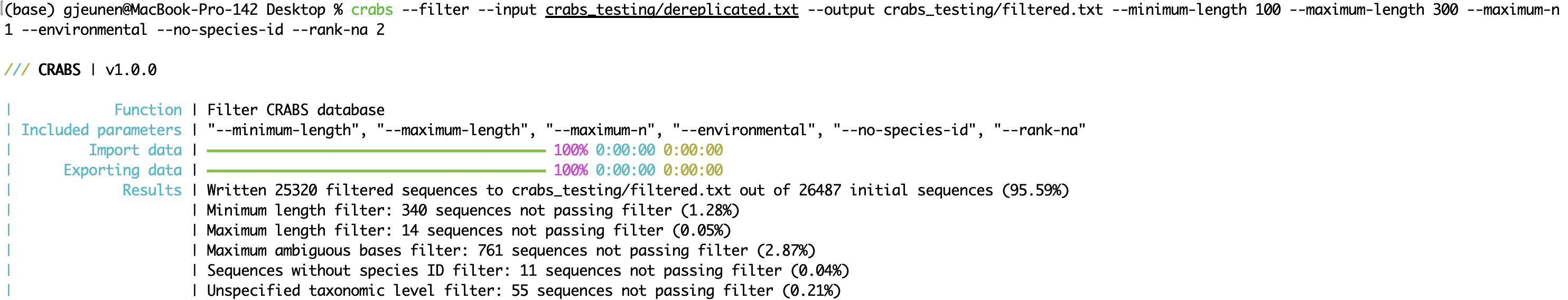
--minimum-length : 데이터베이스에 유지될 앰플리콘의 최소 시퀀스 길이;--maximum-length : 데이터베이스에 보관될 앰플리콘의 최대 시퀀스 길이입니다.--maximum-n : N개 이상의 모호한 염기가 있는 앰플리콘을 폐기합니다( N ).--environmental : 데이터베이스에서 환경 시퀀스를 삭제합니다.--no-species-id : 종 이름을 사용할 수 없는 시퀀스를 삭제합니다.--rank-na : N개 이상의 지정되지 않은 분류 수준이 있는 시퀀스를 삭제합니다. crabs --filter --input crabs_testing/dereplicated.txt --output crabs_testing/filtered.txt --minimum-length 100 --maximum-length 300 --maximum-n 1 --environmental --no-species-id --rank-na 2
--subset CRABS에 통합된 세 번째이자 마지막 큐레이션 방법은 --subset 함수를 사용하여 특정 분류군을 포함(매개변수: --include ) 또는 제외(매개변수: --exclude )하도록 로컬 참조 데이터베이스의 하위 집합을 지정하는 것입니다. 이 기능을 사용하면 연구 질문에 관심이 없는 분류 그룹에서 참조 바코드를 제거할 수 있습니다. 이러한 분류학적 그룹은 프라이머 세트의 잠재적인 비특이적 증폭으로 인해 로컬 참조 데이터베이스에 통합되었을 수 있습니다. --subset 의 또 다른 사용 사례는 알려진 잘못된 시퀀스를 제거하는 것입니다.
기계 학습(IDTAXA) 또는 k-mer 거리(SINTAX)를 기반으로 하는 분류 분류기의 경우 샘플을 채취한 지역에서 발생하는 것으로 알려진 분류군만 포함하고 밀접하게 관련되지 않은 것으로 알려진 종은 제외하여 참조 데이터베이스의 하위 집합을 만드는 것이 도움이 될 수 있습니다. 이러한 분류자의 획득된 분류학적 해상도를 높이고 개선된 분류학적 할당 결과를 얻기 위해 지역에서 발생합니다.
입력 및 출력 파일은 각각 --input 및 --output 매개변수를 사용하여 지정할 수 있습니다. --include 및 --exclude 매개변수는 ; 또는 한 줄에 단일 분류군 이름이 포함된 .txt 파일.
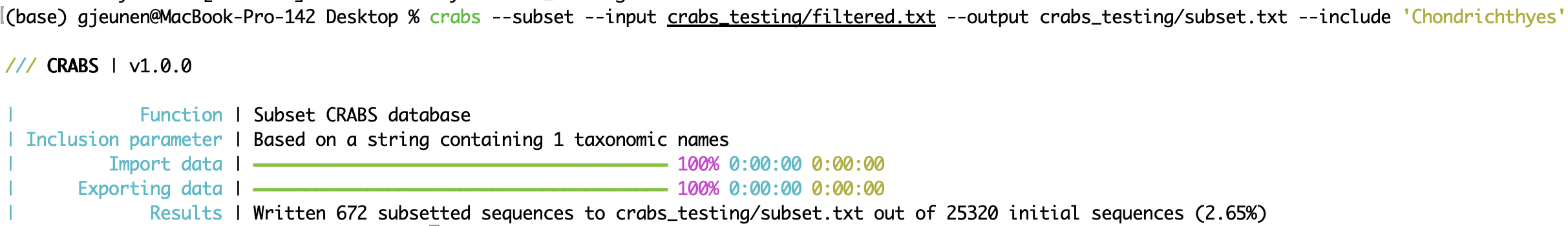
crabs --subset --input crabs_testing/filtered.txt --output crabs_testing/subset.txt --include 'Chondrichthyes'
참조 데이터베이스가 완성되면 메타게놈 데이터에 분류를 할당하는 대부분의 소프트웨어 도구에 필요한 사양을 수용하기 위해 다양한 형식으로 내보낼 수 있습니다. 입력 및 출력 파일은 각각 --input 및 --output 매개변수를 사용하여 지정할 수 있습니다. 버전 v 1.0.0 부터 CRABS는 다음을 포함하여 6개의 서로 다른 분류자(매개변수: --export-format )에 대한 참조 데이터베이스의 형식을 통합합니다.
--export-format 'sintax' : SINTAX 분류자는 USEARCH 및 VSEARCH에 통합됩니다.--export-format 'rdp' : RDP 분류기는 미생물군집 연구에 널리 사용되는 독립 실행형 프로그램입니다.--export-format 'qiime-fasta' 및 --export-format 'qiime-text' : QIIME 및 QIIME2에서 분류 ID를 할당하는 데 사용할 수 있습니다.--export-format 'dada2-species' 및 --export-format 'dada2-taxonomy' : DADA2에서 분류 ID를 할당하는 데 사용할 수 있습니다.--export-format 'idt-fasta' 및 --export-format 'idt-text' : IDTAXA 분류자는 DECIPHER R 패키지에 통합된 기계 학습 알고리즘입니다.--export-format 'blast-notax' : 출력이 분류 ID를 제공하지 않지만 가입 번호를 나열하는 Blastn 및 Megablast에 대한 로컬 Blast Reference 데이터베이스를 만듭니다.--export-format 'blast-tax' : 출력이 분류 ID 및 가입 번호를 모두 제공하는 Blastn 및 Megablast에 대한 로컬 Blast Reference 데이터베이스를 만듭니다. crabs --export --input crabs_testing/subset.txt --output crabs_testing/BLAST_TAX_CHONDRICHTHYES --export-format 'blast-tax'

로컬 참조 데이터베이스를 단일 형식으로 내보내는 동안 (참조 데이터베이스가 여러 파일로 분할되는 분류기를 제외하고, 즉 대부분의 사용자에게는 간단한 루프를 기록 할 수 있습니다. 사용자가 다른 분류 학적 분류기 간의 결과를 비교하려는 경우 여러 형식으로 데이터베이스를 참조하십시오. SINTAX, RDP 및 IDTAXA 형식의 로컬 참조 데이터베이스를 내보내기위한 예는 아래에 제공됩니다.
for format in sintax.fasta rdp.fasta idt-fasta.fasta idt-text.txt; do crabs --export --input crabs_testing/subset.txt --output crabs_testing/chondrichthyes_${format} --export-format ${format%%.*}; done

참조 데이터베이스가 완료되면 Crabs는 (i) --diversity-figure , (ii) --amplicon-length-figure . iii) --phylogenetic-tree , (iv) --amplification-efficiency-figure 및 (v) --completeness-table .
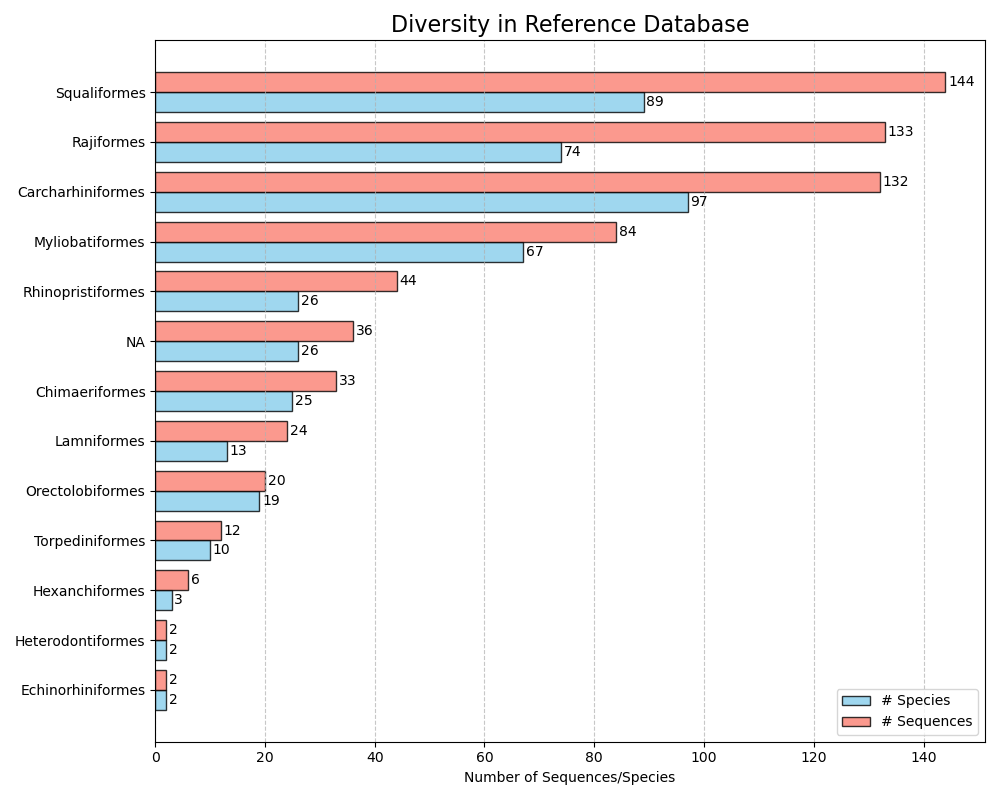
--diversity-figure --diversity-figure 함수는 참조 데이터베이스의 각 분류 학적 그룹에 대해 종 수 (파란색) 및 시퀀스 수 (주황색)를 가진 수평 막대 플롯을 생성합니다. 사용자는 분류 학적 순위를 지정하여 참조 데이터베이스를 --tax-level 매개 변수로 분할 할 수 있습니다. 세금 수준은 --import 기능 중에 나타난 순위 수입니다. 예를 들어, --ranks 'superkingdom;phylum;class;order;family;genus;species' --import 슈퍼 킹을 기반으로 한 import 분할 중에 사용 되었다면 --tax-level 1 , phylum = --tax-level 2 , class = --tax-level 3 등. 크랩 형식의 입력 파일은 --input 매개 변수를 사용하여 지정할 수 있습니다. .png 형식의 그림은 --output 매개 변수를 사용하여 지정할 수있는 출력 파일에 기록됩니다.
crabs --diversity-figure --input crabs_testing/subset.txt --output crabs_testing/diversity-figure.png --tax-level 4
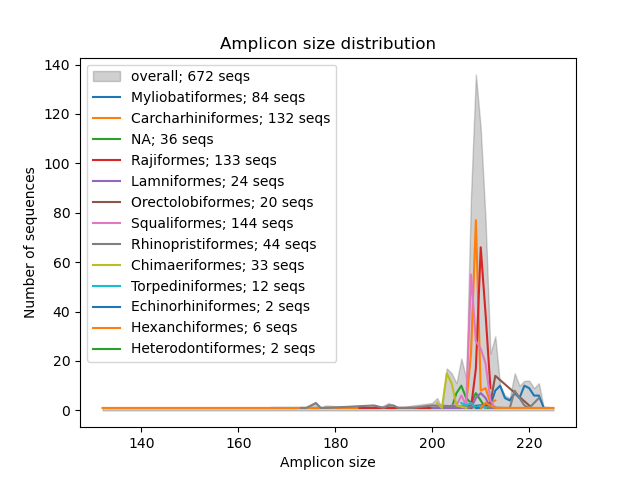
--amplicon-length-figure --amplicon-length-figure 함수는 앰플 리콘 길이의 범위를 표시하는 라인 그래프를 생성합니다. 참조 데이터베이스의 모든 서열에 걸친 앰플 리콘 길이의 전체 범위는 그늘진 회색으로 표시되는 반면, 분류 학적 그룹 (매개 변수 : --tax-level ) 당 분할 결과는 색상 선으로 오버레이됩니다. 또한, 범례는 각 분류 그룹에 할당 된 시퀀스 수와 참조 데이터베이스의 총 시퀀스 수를 표시합니다. 크랩 형식의 입력 파일은 --input 매개 변수를 사용하여 지정할 수 있습니다. .png 형식의 그림은 --output 매개 변수를 사용하여 지정할 수있는 출력 파일에 기록됩니다.
crabs --amplicon-length-figure --input crabs_testing/subset.txt --output crabs_testing/amplicon-length-figure.png --tax-level 4
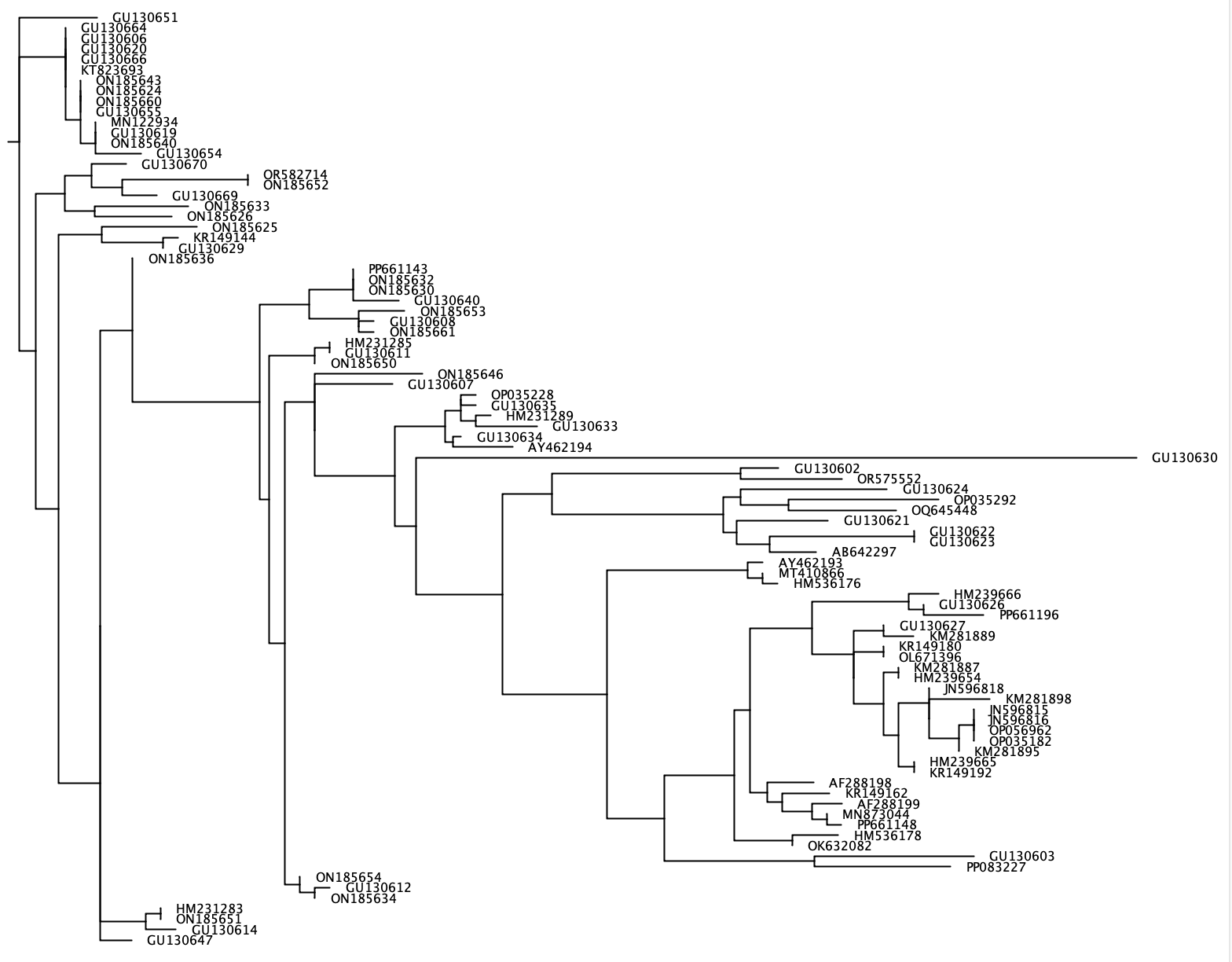
--phylogenetic-tree --phylogenetic-tree 기능은 관심 종 목록에 대한 계통 발생 트리를 생성합니다. 이 관심 종의 목록은 --species 매개 변수를 사용하여 가져올 수 있으며 각 줄에 단일 종 이름을 가진 + 또는 .txt 파일로 구분 된 입력 문자열로 구성됩니다. 관심있는 각 종에 대해, 서열은 사용자 정의 분류 학적 순위 (매개 변수 : --tax-level )를 관심있는 종과 공유하는 참조 데이터베이스에서 추출 될 것이다. 게는 clustalw2 v 2.1을 사용하여 추출 된 모든 서열의 정렬을 생성하고 Fasttree를 사용하여 이웃 결합 계통 발생 트리를 생성합니다. Newick 형식 의이 계통 발생 트리는 --output 매개 변수를 사용하여 출력 파일에 기록되며 Figtree 또는 Geneious와 같은 소프트웨어 프로그램에서 시각화 될 수 있습니다. 관심있는 각 종에 대해 별도의 계통 발생 트리가 생성되므로 --output 일반 파일 이름으로 가져 오는 반면, 정확한 출력 파일에는이 일반 이름과 '_species_name.tree'가 포함됩니다.
crabs --phylogenetic-tree --input crabs_testing/subset.txt --output crabs_testing/phylo --tax-level 4 --species 'Carcharodon carcharias+Squalus acanthias'
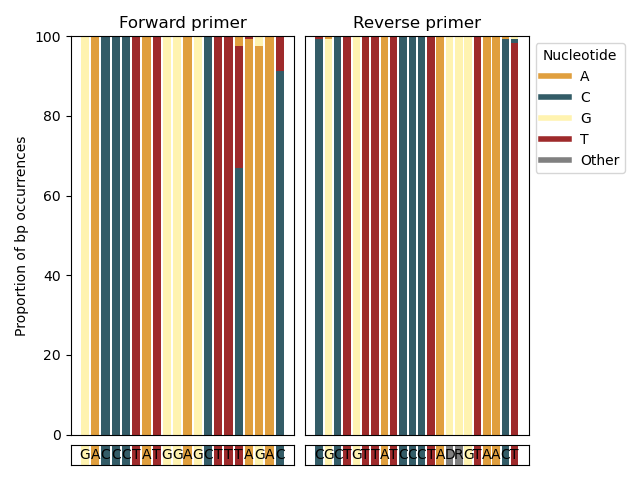
--amplification-efficiency-figure --amplification-efficiency-figure 함수는 막대 그래프를 생성하여 사용자 지정 분류 그룹에 대한 프라이머 바인딩 영역에서 기본 쌍 발생의 비율을 표시함으로써, 안내 및 역방향 프라이머-결합 영역에서 장소를 시각화하여 불일치합니다. 분류 학적 관심 그룹에서 발생할 수 있으며, 증폭 효율에 영향을 줄 수 있습니다. --amplification-efficiency-figure 함수는 --amplicons 매개 변수를 사용하여 최종 Crabs 형식의 참조 데이터베이스를 입력으로 사용합니다. 입력 파일의 각 시퀀스에 대한 프라이머 바인딩 영역에 대한 정보를 찾으려면, 가져 오기 후 처음 다운로드 된 시퀀스는 --input 매개 변수를 사용하여 제공되어야합니다. 전방 및 리버스 프라이머 서열 (5 '-3'방향)은 --forward 및 --reverse 매개 변수를 사용하여 제공됩니다. 분류 학적 관심 그룹의 이름은 --tax-group 매개 변수를 사용하여 제공 될 수 있으며 입력 파일에 통합 된 모든 분류 수준으로 설정할 수 있습니다. 마지막으로, .png 형식의 그림은 --output 매개 변수로 지정된 출력 파일에 기록됩니다.
crabs --amplification-efficiency-figure --input crabs_testing/merged.txt --amplicons crabs_testing/subset.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT --output crabs_testing/amplification-efficiency.png --tax-group Carcharhiniformes
--completeness-table --completeness-table 함수는 관심 종 목록에 대한 정보를 사용하여 탭으로 변형 된 테이블 (매개 변수 : --output )을 출력합니다. 이 관심 종의 목록은 --species 매개 변수를 사용하여 가져올 수 있으며 각 줄에 단일 종 이름을 가진 + 또는 .txt 파일로 구분 된 입력 문자열로 구성됩니다. 각각 --names 및 --nodes 매개 변수를 사용하여 --download-taxonomy 함수를 사용하여 다운로드 한 ' names.dmp '및 ' nodes.dmp '파일을 사용하여 각 관심 종에 대해 분류 계보가 생성됩니다. 출력 테이블에는 다음 정보를 제공하는 10 개의 열이 있습니다.
crabs --completeness-table --input crabs_testing/subset.txt --output crabs_testing/completeness.txt --names crabs_testing/names.dmp --nodes crabs_testing/nodes.dmp --species 'Carcharodon carcharias+Squalus acanthias'
crabs --version v 1.0.6 : 버그 수정 -> --import 동안 대담한 헤더의 개선 된 구문 분석.crabs --version v 1.0.5 : 버그 수정 -> Blast+ 소프트웨어에 필요한 경우 Blast 데이터베이스를 구축 할 때 SEQ ID에 길이 제한 사항을 추가했습니다.crabs --version v 1.0.4 : 추가 정보-> --pairwise-global-alignment --coverage --percent-identity 에 대한 값 입력에 대한 올바른 정보 제공-crabs --version v 1.0.3 : 버그 수정 -> NCBI 서버 응답 확인 분석을 중단하기 전에 3 번 확인하십시오.crabs --version v 1.0.2 : 버그 수정 -> 분석 후 0 시퀀스가 반환 될 때보고 할 수 있습니다.crabs --version v 1.0.1 : 버그 수정 -> 성공적인 구축 NCBI 쿼리를 사용하여 --species 매개 변수를 사용합니다.

