BindCraft
v1.2.0
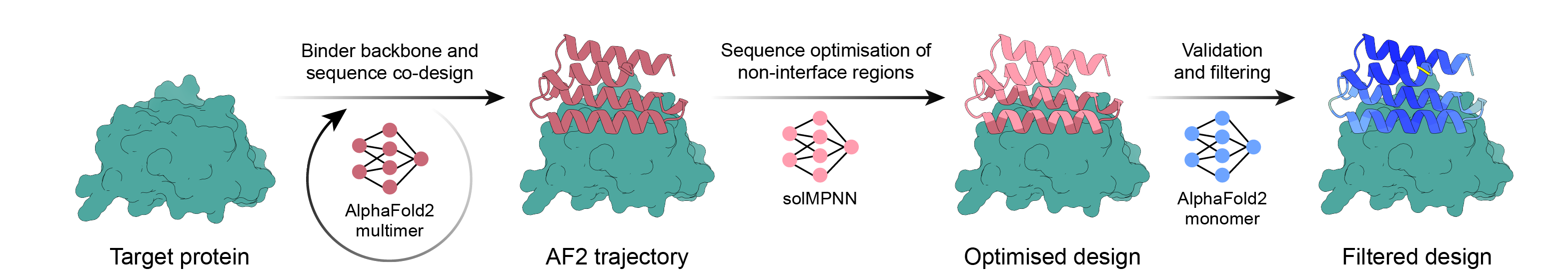
AlphaFold2 역전파, MPNN 및 PyRosetta를 사용한 간단한 바인더 설계 파이프라인. 대상을 선택하고 스크립트가 나머지 작업을 수행하도록 하고 주문할 디자인이 충분하면 완료하세요!
BindCraft 사전 인쇄 링크
먼저 이 저장소를 복제해야 합니다. [install_folder]를 설치하려는 경로로 바꾸세요.
git clone https://github.com/martinpacesa/BindCraft [install_folder]
CD를 사용하여 설치 폴더로 이동하고 설치 코드를 실행합니다. BindCraft를 실행하려면 CUDA 호환 Nvidia 그래픽 카드가 필요합니다. Cuda 설정에서 그래픽 카드와 호환되는 CUDA 버전을 지정하세요(예: '11.8'). 확실하지 않은 경우 공백으로 두십시오. 그러나 설치 시 잘못된 버전을 선택하여 오류가 발생할 수 있습니다. pkg_manager 에서 'mamba' 또는 'conda'를 사용할지 지정합니다. 공백으로 두면 기본적으로 'conda'가 사용됩니다.
참고: 이 설치 스크립트는 상업적 목적으로 라이센스가 필요한 PyRosetta를 설치합니다.
bash install_bindcraft.sh --cuda '12.4' --pkg_manager 'conda'
항상 입력 대상 PDB를 가능한 가장 작은 크기로 자르도록 노력하세요! 바인더 생성 속도를 크게 높이고 GPU 메모리 요구 사항을 최소화합니다.
허용되는 바인더를 확인하려면 최소한 수백 개의 궤적을 실행할 준비를 하십시오. 어려운 목표의 경우 수천 개가 될 수도 있습니다.
스크립트를 로컬에서 실행하려면 먼저 settings_target 폴더에서 대상 .json 파일을 구성해야 합니다. json 파일에는 다음 설정이 있습니다.
design_path -> path where to save designs and statistics
binder_name -> what to prefix your designed binder files with
starting_pdb -> the path to the PDB of your target protein
chains -> which chains to target in your protein, rest will be ignored
target_hotspot_residues -> which position to target for binder design, for example `1,2-10` or chain specific `A1-10,B1-20` or entire chains `A`, set to null if you want AF2 to select binding site; better to select multiple target residues or a small patch to reduce search space for binder
lengths -> range of binder lengths to design
number_of_final_designs -> how many designs that pass all filters to aim for, script will stop if this many are reached
그런 다음 바인더 디자인 스크립트를 실행합니다.
sbatch ./bindcraft.slurm --settings './settings_target/PDL1.json' --filters './settings_filters/default_filters.json' --advanced './settings_advanced/default_4stage_multimer.json'
설정 플래그는 위에서 설정한 대상 .json을 가리켜야 합니다. 필터 플래그는 디자인 필터가 지정된 json을 가리킵니다(기본값은 ./filters/default_filters.json). 고급 플래그는 고급 설정을 가리킵니다(기본값은 ./advanced_settings/default_4stage_multimer.json). 필터 및 고급 설정 플래그를 생략하면 자동으로 기본값을 가리킵니다.
또는 컴퓨터가 SLURM을 지원하지 않는 경우 conda에서 환경을 활성화하고 Python 코드를 실행하여 코드를 직접 실행할 수 있습니다.
conda activate BindCraft
cd /path/to/bindcraft/folder/
python -u ./bindcraft.py --settings './settings_target/PDL1.json' --filters './settings_filters/default_filters.json' --advanced './settings_advanced/default_4stage_multimer.json'
모든 필터를 통과한 최소 100개의 최종 디자인을 생성한 다음 실험적 특성화를 위해 상위 5~20개를 주문하는 것이 좋습니다. 높은 친화력 바인더가 필요한 경우 순위 지정에 사용되는 ipTM 측정항목은 친화력에 대한 좋은 예측 변수는 아니지만 결합에 대한 좋은 이진 예측 변수인 것으로 나타났기 때문에 더 많이 선별하는 것이 좋습니다.
다음은 개별 필터 및 고급 설정에 대한 설명입니다.
디자인 프로세스를 제어하는 고급 설정은 다음과 같습니다.
omit_AAs -> which amino acids to exclude from design (note: they can still occur if no other options are possible in the position)
force_reject_AA -> whether to force reject design if it contains any amino acids specified in omit_AAs
design_algorithm -> which design algorithm for the trajecory to use, the currently implemented algorithms are below
use_multimer_design -> whether to use AF2-ptm or AF2-multimer for binder design; the other model will be used for validation then
num_recycles_design -> how many recycles of AF2 for design
num_recycles_validation -> how many recycles of AF2 use for structure prediction and validation
sample_models = True -> whether to randomly sample parameters from AF2 models, recommended to avoid overfitting
rm_template_seq_design -> remove target template sequence for design (increases target flexibility)
rm_template_seq_predict -> remove target template sequence for reprediction (increases target flexibility)
rm_template_sc_design -> remove sidechains from target template for design
rm_template_sc_predict -> remove sidechains from target template for reprediction
# Design iterations
soft_iterations -> number of soft iterations (all amino acids considered at all positions)
temporary_iterations -> number of temporary iterations (softmax, most probable amino acids considered at all positions)
hard_iterations -> number of hard iterations (one hot encoding, single amino acids considered at all positions)
greedy_iterations -> number of iterations to sample random mutations from PSSM that reduce loss
greedy_percentage -> What percentage of protein length to mutate during each greedy iteration
# Design weights, higher value puts more weight on optimising the parameter.
weights_plddt -> Design weight - pLDDT of designed chain
weights_pae_intra -> Design weight - PAE within designed chain
weights_pae_inter -> Design weight - PAE between chains
weights_con_intra -> Design weight - maximise number of contacts within designed chain
weights_con_inter -> Design weight - maximise number of contacts between chains
intra_contact_distance -> Cbeta-Cbeta cutoff distance for contacts within the binder
inter_contact_distance -> Cbeta-Cbeta cutoff distance for contacts between binder and target
intra_contact_number -> how many contacts each contact esidue should make within a chain, excluding immediate neighbours
inter_contact_number -> how many contacts each contact residue should make between chains
weights_helicity -> Design weight - helix propensity of the design, Default 0, negative values bias towards beta sheets
random_helicity -> whether to randomly sample helicity weights for trajectories, from -1 to 1
# Additional losses
use_i_ptm_loss -> Use i_ptm loss to optimise for interface pTM score?
weights_iptm -> Design weight - i_ptm between chains
use_rg_loss -> use radius of gyration loss?
weights_rg -> Design weight - radius of gyration weight for binder
use_termini_distance_loss -> Try to minimise distance between N- and C-terminus of binder? Helpful for grafting
weights_termini_loss -> Design weight - N- and C-terminus distance minimisation weight of binder
# MPNN settings
mpnn_fix_interface -> whether to fix the interface designed in the starting trajectory
num_seqs -> number of MPNN generated sequences to sample and predict per binder
max_mpnn_sequences -> how many maximum MPNN sequences per trajectory to save if several pass filters
max_tm-score_filter -> filter out final lower ranking designs by this TM score cut off relative to all passing designs
max_seq-similarity_filter -> filter out final lower ranking designs by this sequence similarity cut off relative to all passing designs
sampling_temp = 0.1 -> sampling temperature for amino acids, T=0.0 means taking argmax, T>>1.0 means sampling randomly.")
# MPNN settings - advanced
sample_seq_parallel -> how many sequences to sample in parallel, reduce if running out of memory
backbone_noise -> backbone noise during sampling, 0.00-0.02 are good values
model_path -> path to the MPNN model weights
mpnn_weights -> whether to use "original" mpnn weights or "soluble" weights
save_mpnn_fasta -> whether to save MPNN sequences as fasta files, normally not needed as the sequence is also in the CSV file
# AF2 design settings - advanced
num_recycles_design -> how many recycles of AF2 for design
num_recycles_validation -> how many recycles of AF2 use for structure prediction and validation
optimise_beta -> optimise predictions if beta sheeted trajectory detected?
optimise_beta_extra_soft -> how many extra soft iterations to add if beta sheets detected
optimise_beta_extra_temp -> how many extra temporary iterations to add if beta sheets detected
optimise_beta_recycles_design -> how many recycles to do during design if beta sheets detected
optimise_beta_recycles_valid -> how many recycles to do during reprediction if beta sheets detected
# Optimise script
remove_unrelaxed_trajectory -> remove the PDB files of unrelaxed designed trajectories, relaxed PDBs are retained
remove_unrelaxed_complex -> remove the PDB files of unrelaxed predicted MPNN-optimised complexes, relaxed PDBs are retained
remove_binder_monomer -> remove the PDB files of predicted binder monomers after scoring to save space
zip_animations -> at the end, zip Animations trajectory folder to save space
zip_plots -> at the end, zip Plots trajectory folder to save space
save_trajectory_pickle -> save pickle file of the generated trajectory, careful, takes up a lot of storage space!
max_trajectories -> how many maximum trajectories to generate, for benchmarking
acceptance_rate -> what fraction of trajectories should yield designs passing the filters, if the proportion of successful designs is less than this fraction then the script will stop and you should adjust your design weights
start_monitoring -> after what number of trajectories should we start monitoring acceptance_rate, do not set too low, could terminate prematurely
# debug settings
enable_mpnn = True -> whether to enable MPNN design
enable_rejection_check -> enable rejection rate check
디자인을 필터링하는 기능은 다음과 같습니다. 일부 기능을 사용하지 않으려면 임계값으로 null을 설정하면 됩니다. 더 높은 옵션은 임계값보다 높은 값을 유지해야 하는지(true) 아니면 더 낮게 유지해야 하는지(false) 여부를 나타냅니다. N_으로 시작하는 기능은 각 AlphaFold 모델별 통계에 해당하며, 평균은 예측된 모든 모델에 대한 것입니다.
MPNN_score -> MPNN sequence score, generally not recommended as it depends on protein
MPNN_seq_recovery -> MPNN sequence recovery of original trajectory
pLDDT -> pLDDT confidence score of AF2 complex prediction, normalised to 0-1
pTM -> pTM confidence score of AF2 complex prediction, normalised to 0-1
i_pTM -> interface pTM confidence score of AF2 complex prediction, normalised to 0-1
pAE -> predicted alignment error of AF2 complex prediction, normalised compared AF2 by n/31 to 0-1
i_pAE -> predicted interface alignment error of AF2 complex prediction, normalised compared AF2 by n/31 to 0-1
i_pLDDT -> interface pLDDT confidence score of AF2 complex prediction, normalised to 0-1
ss_pLDDT -> secondary structure pLDDT confidence score of AF2 complex prediction, normalised to 0-1
Unrelaxed_Clashes -> number of interface clashes before relaxation
Relaxed_Clashes -> number of interface clashes after relaxation
Binder_Energy_Score -> Rosetta energy score for binder alone
Surface_Hydrophobicity -> surface hydrophobicity fraction for binder
ShapeComplementarity -> interface shape complementarity
PackStat -> interface packstat rosetta score
dG -> interface rosetta dG energy
dSASA -> interface delta SASA (size)
dG/dSASA -> interface energy divided by interface size
Interface_SASA_% -> Fraction of binder surface covered by the interface
Interface_Hydrophobicity -> Interface hydrophobicity fraction of binder interface
n_InterfaceResidues -> number of interface residues
n_InterfaceHbonds -> number of hydrogen bonds at the interface
InterfaceHbondsPercentage -> number of hydrogen bonds compared to interface size
n_InterfaceUnsatHbonds -> number of unsatisfied buried hydrogen bonds at the interface
InterfaceUnsatHbondsPercentage -> number of unsatisfied buried hydrogen bonds compared to interface size
Interface_Helix% -> proportion of alfa helices at the interface
Interface_BetaSheet% -> proportion of beta sheets at the interface
Interface_Loop% -> proportion of loops at the interface
Binder_Helix% -> proportion of alfa helices in the binder structure
Binder_BetaSheet% -> proportion of beta sheets in the binder structure
Binder_Loop% -> proportion of loops in the binder structure
InterfaceAAs -> number of amino acids of each type at the interface
HotspotRMSD -> unaligned RMSD of binder compared to original trajectory, in other words how far is binder in the repredicted complex from the original binding site
Target_RMSD -> RMSD of target predicted in context of the designed binder compared to input PDB
Binder_pLDDT -> pLDDT confidence score of binder predicted alone
Binder_pTM -> pTM confidence score of binder predicted alone
Binder_pAE -> predicted alignment error of binder predicted alone
Binder_RMSD -> RMSD of binder predicted alone compared to original trajectory
코딩과 아이디어 논의에 도움을 준 Lennart Nickel, Yehlin Cho, Casper Goverde 및 Sergey Ovchinnikov에게 감사드립니다. 이 저장소는 다음의 코드를 사용합니다.


