nucleotide transformer
1.0.0
InstaDeep Github 저장소에 오신 것을 환영합니다.
우리는 이러한 작업을 오픈 소스로 제공하고 커뮤니티에 이러한 9가지 게놈 언어 모델과 2가지 분할 모델에 대한 코드 및 사전 훈련된 가중치에 대한 액세스를 제공하게 되어 기쁘게 생각합니다. nucleotide transformer 프로젝트의 모델은 Nvidia 및 TUM과 협력하여 개발되었으며, 모델은 Cambridge-1의 DGX A100 노드에서 훈련되었습니다. Agro nucleotide transformer 프로젝트의 모델은 Google과 공동으로 개발되었으며 모델은 TPU-v4 가속기에서 훈련되었습니다.
전반적으로, 우리의 작업은 언어 기본 모델의 사전 훈련 및 적용과 관련된 새로운 통찰력을 제공할 뿐만 아니라 이를 백본 인코더로 사용하는 모델의 훈련과 현장에서 적용할 수 있는 충분한 기회를 통해 유전체학에 대한 새로운 통찰력을 제공합니다.
이 저장소에서는 다음을 찾을 수 있습니다.
다른 접근법과 비교하여, 우리의 모델은 단일 참조 게놈의 정보를 통합할 뿐만 아니라 3,200개 이상의 다양한 인간 게놈뿐만 아니라 모델 및 비모델 유기체를 포함한 광범위한 종의 850개 게놈의 DNA 서열을 활용합니다. 강력하고 광범위한 평가를 통해 우리는 이러한 대형 모델이 기존 방법에 비해 매우 정확한 분자 표현형 예측을 제공한다는 것을 보여줍니다.
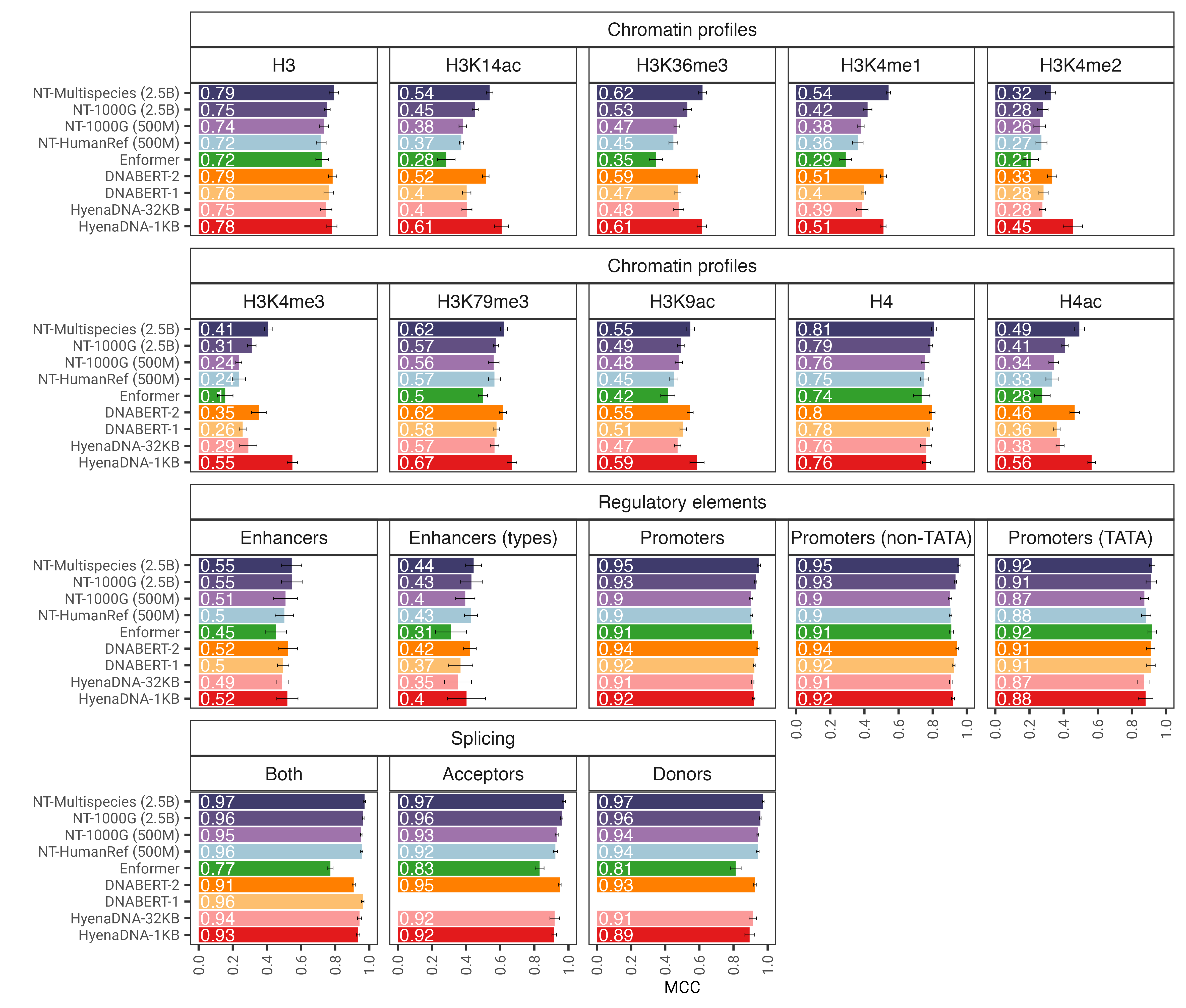
그림 1: nucleotide transformer 모델은 미세 조정 후 다양한 유전체학 작업을 정확하게 예측합니다. 미세 조정된 변압기 모델에 대한 다운스트림 작업 전반의 성능 결과를 보여줍니다. 오차 막대는 10겹 교차 검증에서 파생된 2개의 SD를 나타냅니다.
이 연구에서 우리는 작물 종에 주로 초점을 맞춘 48개 식물 종의 참조 게놈에 대해 훈련된 새로운 기초 대형 언어 모델을 제시합니다. 우리는 규제 기능, RNA 처리 및 유전자 발현에 이르는 여러 예측 작업에 걸쳐 AgroNT의 성능을 평가하고 AgroNT가 최첨단 성능을 얻을 수 있음을 보여줍니다.
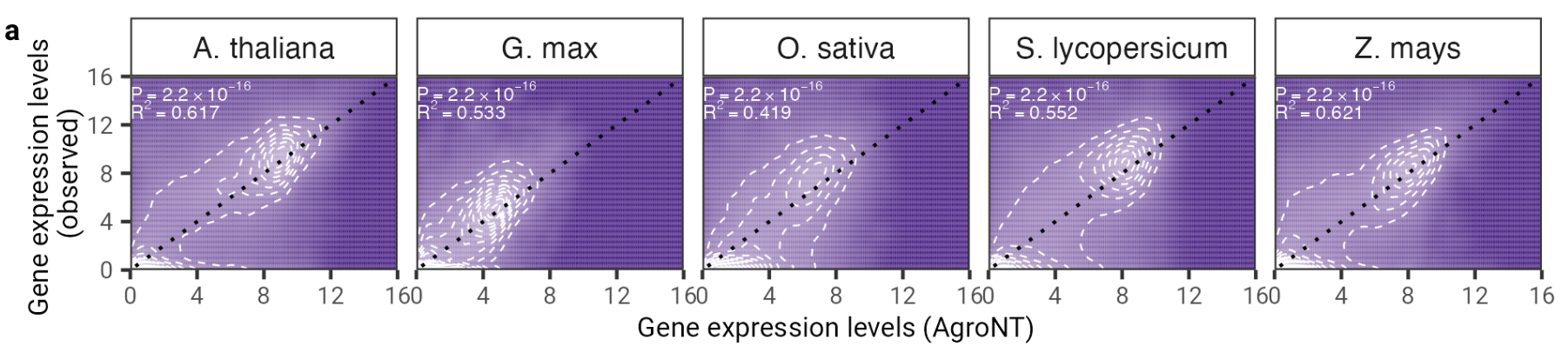
그림 2: AgroNT는 다양한 식물 종에 걸쳐 유전자 발현 예측을 제공합니다. 모든 조직에 걸쳐 홀드아웃 유전자에 대한 유전자 발현 예측은 관찰된 유전자 발현 수준과 상관관계가 있습니다. 선형 모델의 결정 계수(R 2 )와 예측 값과 관측 값 사이의 관련 P 값이 표시됩니다.
코드와 사전 학습된 모델을 사용하려면 다음을 수행하세요.
pip install . .그런 다음 단 몇 줄의 코드만으로 9가지 모델 중 하나를 다운로드하고 추론을 수행할 수 있습니다.
import haiku as hk
import jax
import jax . numpy as jnp
from nucleotide_transformer . pretrained import get_pretrained_model
# Get pretrained model
parameters , forward_fn , tokenizer , config = get_pretrained_model (
model_name = "500M_human_ref" ,
embeddings_layers_to_save = ( 20 ,),
max_positions = 32 ,
)
forward_fn = hk . transform ( forward_fn )
# Get data and tokenize it
sequences = [ "ATTCCGATTCCGATTCCG" , "ATTTCTCTCTCTCTCTGAGATCGATCGATCGAT" ]
tokens_ids = [ b [ 1 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens_str = [ b [ 0 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens = jnp . asarray ( tokens_ids , dtype = jnp . int32 )
# Initialize random key
random_key = jax . random . PRNGKey ( 0 )
# Infer
outs = forward_fn . apply ( parameters , random_key , tokens )
# Get embeddings at layer 20
print ( outs [ "embeddings_20" ]. shape )지원되는 모델 이름은 다음과 같습니다.
또한 모델을 실행하고 Google Colab에서 더 많은 예제 코드를 찾을 수도 있습니다.
Jax 덕분에 코드는 GPU와 TPU 모두에서 실행됩니다!
두 번째 버전의 nucleotide transformer v2 모델에는 보다 효율적인 것으로 입증된 일련의 아키텍처 변경이 포함되어 있습니다. 학습된 위치 임베딩을 사용하는 대신 각 주의 계층에서 사용되는 로터리 임베딩과 편견 없는 스위시 활성화가 있는 Gated Linear Unit을 사용합니다. 이러한 개선된 모델은 또한 최대 2,048개의 토큰 시퀀스를 허용하여 12kbp의 더 긴 컨텍스트 창으로 이어집니다. Chinchilla 스케일링 법칙에서 영감을 받아 v1 모델(300B 토큰)과 비교하여 더 오랜 기간(50M 및 100M 모델의 경우 300B 토큰, 250M 및 500M 모델의 경우 1T 토큰) 동안 다중 종 데이터 세트에서 NT-v2 모델을 교육했습니다. 네 가지 모델 모두에 해당).
변환기 레이어는 1-인덱싱됩니다. 즉, model_name="500M_human_ref" 및 embeddings_layers_to_save=(1, 20,) 인수를 사용하여 get_pretrained_model 호출하면 첫 번째 및 20번째 변환기 레이어 이후의 임베딩이 추출됩니다. Roberta LM 헤드를 사용하는 변환기의 경우 마지막 변환기 블록 이후가 아닌 LM 헤드의 첫 번째 레이어 표준 이후에 최종 임베딩을 추출하는 것이 일반적입니다. 따라서 get_pretrained_model 다음 인수 embeddings_layers_to_save=(24,) 와 함께 호출되면 임베딩은 최종 변환기 레이어 이후가 아니라 LM 헤드의 첫 번째 레이어 표준 이후에 추출됩니다.
SegmentNT 모델은 언어 모델 헤드를 제거하고 1차원 U-Net 분할 헤드로 대체한 NT( nucleotide transformer ) 변환기를 활용하여 단일 뉴클레오티드 분해능에서 시퀀스의 여러 유형의 게놈 요소 위치를 예측합니다. 우리는 최대 30kb의 입력 시퀀스에서 인간 유전체학 요소의 14가지 클래스에 대한 두 가지 모델 변형을 제시합니다. 여기에는 유전자(단백질 코딩 유전자, lncRNA, 5'UTR, 3'UTR, 엑손, 인트론, 스플라이스 수용체 및 공여자 부위)와 조절(polyA 신호, 조직 불변 및 조직 특이적 프로모터 및 인핸서, CTCF 결합)이 포함됩니다. 사이트) 요소. SegmentNT는 사전 훈련된 NT 가중치의 이점을 활용하여 최첨단 U-Net 분할 아키텍처에 비해 뛰어난 성능을 달성하고 최대 50kbp의 제로샷 일반화를 보여줍니다.
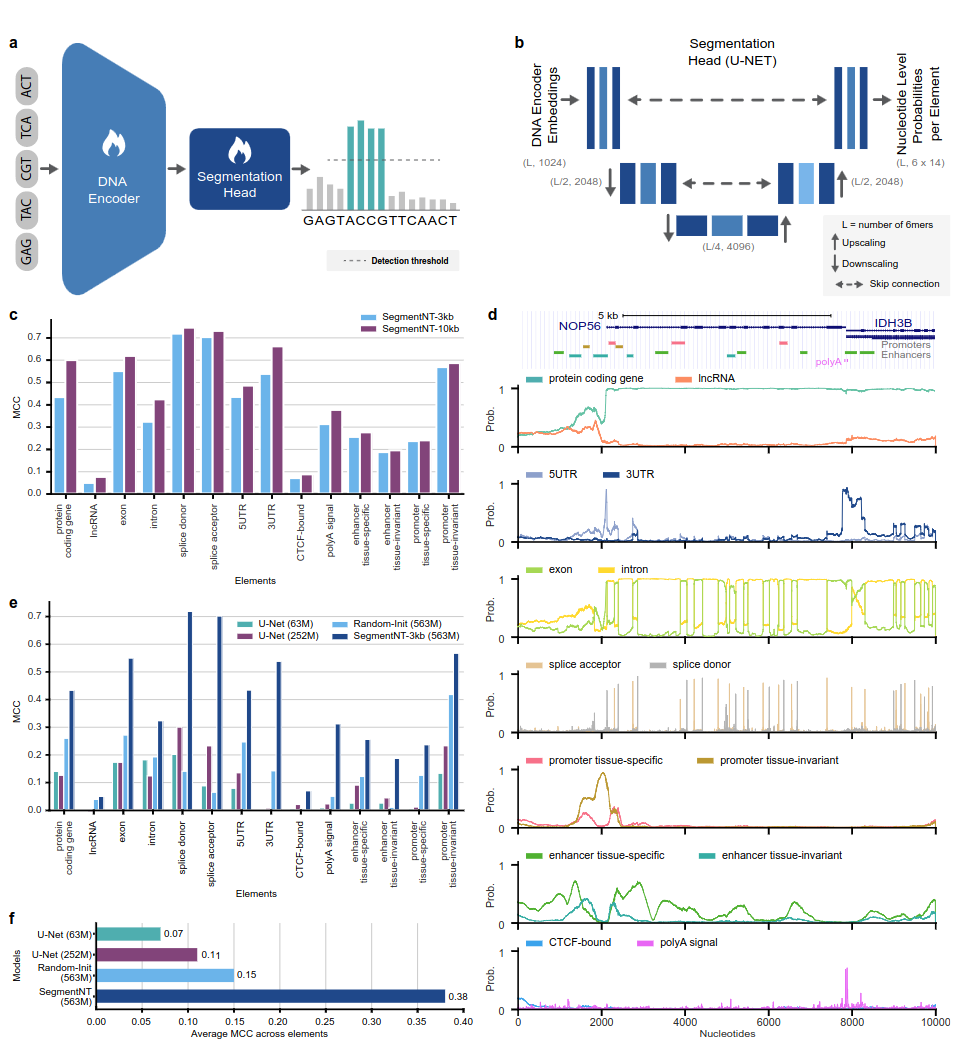
그림 1: SegmentNT는 뉴클레오티드 분해능에서 게놈 요소를 위치화합니다.
코드와 사전 학습된 모델을 사용하려면 다음을 수행하세요.
pip install . .그런 다음 단 몇 줄의 코드만으로 당사 모델의 시퀀스를 다운로드하고 추론할 수 있습니다.
rescaling factor 훈련 중에 사용된 요소로 설정됩니다. 30kbp에서 50kbp 사이의 시퀀스를 추론해야 하는 경우 get_pretrained_segment_nt_model 함수에서 rescaling_factor 인수를 rescaling_factor = max_num_nucleotides / max_num_tokens_nt 값으로 전달해야 합니다. 여기서 num_dna_tokens_inference 는 추론 시 토큰 수입니다(예: 40008 염기 시퀀스의 경우 6669). 쌍) 및 max_num_tokens_nt 는 백본 뉴클레오티드 변환기가 훈련된 최대 토큰 수입니다(예: 2048 .
? 노트북 examples/inference_segment_nt.ipynb 50kb 시퀀스를 추론하고 논문의 그림 3을 재현할 확률을 그리는 방법을 보여줍니다.
? SegmentNT 모델은 각 뉴클레오티드를 6-mer로 토큰화해야 하기 때문에 입력 시퀀스의 "N"을 처리하지 않습니다. 이는 하나 또는 여러 개의 "N" 염기쌍을 포함하는 시퀀스를 사용할 때 그럴 수 없습니다.
import haiku as hk
import jax
import jax . numpy as jnp
from nucleotide_transformer . pretrained import get_pretrained_segment_nt_model
# Initialize CPU as default JAX device. This makes the code robust to memory leakage on
# the devices.
jax . config . update ( "jax_platform_name" , "cpu" )
backend = "cpu"
devices = jax . devices ( backend )
num_devices = len ( devices )
print ( f"Devices found: { devices } " )
# The number of DNA tokens (excluding the CLS token prepended) needs to be dividible by
# 2 to the power of the number of downsampling block, i.e 4.
max_num_nucleotides = 8
assert max_num_nucleotides % 4 == 0 , (
"The number of DNA tokens (excluding the CLS token prepended) needs to be dividible by"
"2 to the power of the number of downsampling block, i.e 4." )
parameters , forward_fn , tokenizer , config = get_pretrained_segment_nt_model (
model_name = "segment_nt" ,
embeddings_layers_to_save = ( 29 ,),
attention_maps_to_save = (( 1 , 4 ), ( 7 , 10 )),
max_positions = max_num_nucleotides + 1 ,
)
forward_fn = hk . transform ( forward_fn )
apply_fn = jax . pmap ( forward_fn . apply , devices = devices , donate_argnums = ( 0 ,))
# Get data and tokenize it
sequences = [ "ATTCCGATTCCGATTCCAACGGATTATTCCGATTAACCGATTCCAATT" , "ATTTCTCTCTCTCTCTGAGATCGATGATTTCTCTCTCATCGAACTATG" ]
tokens_ids = [ b [ 1 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens_str = [ b [ 0 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens = jnp . asarray ( tokens_ids , dtype = jnp . int32 )
random_key = jax . random . PRNGKey ( seed = 0 )
keys = jax . device_put_replicated ( random_key , devices = devices )
parameters = jax . device_put_replicated ( parameters , devices = devices )
tokens = jax . device_put_replicated ( tokens , devices = devices )
# Infer on the sequence
outs = apply_fn ( parameters , keys , tokens )
# Obtain the logits over the genomic features
logits = outs [ "logits" ]
# Transform them in probabilities
probabilities = jnp . asarray ( jax . nn . softmax ( logits , axis = - 1 ))[..., - 1 ]
print ( f"Probabilities shape: { probabilities . shape } " )
print ( f"Features inferred: { config . features } " )
# Get probabilities associated with intron
idx_intron = config . features . index ( "intron" )
probabilities_intron = probabilities [..., idx_intron ]
print ( f"Intron probabilities shape: { probabilities_intron . shape } " )지원되는 모델 이름은 다음과 같습니다.
Jax 덕분에 코드는 GPU와 TPU 모두에서 실행됩니다!
모델은 시퀀스 시작 부분에 자동으로 추가되는 <CLS> 토큰을 포함하여 최대 1000개의 토큰 길이의 시퀀스에 대해 학습됩니다. 토크나이저는 문자 "A", "C", "G" 및 "T"를 6mer로 그룹화하여 왼쪽에서 오른쪽으로 토큰화를 시작합니다. "N" 문자는 k-mer 내부에 그룹화되지 않도록 선택되므로 토크나이저가 "N"을 만날 때마다 또는 시퀀스의 뉴클레오티드 수가 6의 배수가 아닌 경우 그룹화하지 않고 뉴클레오티드를 토큰화합니다. 그들을. 예는 다음과 같습니다.
dna_sequence_1 = "ACGTGTACGTGCACGGACGACTAGTCAGCA"
tokenized_dna_sequence_1 = [ < CLS > , < ACGTGT > , < ACGTGC > , < ACGGAC > , < GACTAG > , < TCAGCA > ]
dna_sequence_2 = "ACGTGTACNTGCACGGANCGACTAGTCTGA"
tokenized_dna_sequence_2 = [ < CLS > , < ACGTGT > , < A > , < C > , < N > , < TGCACG > , < G > , < A > , < N > , < CGACTA > , < GTCTGA > ]따라서 모든 v1 및 v2 변환기는 내부에 "N"이 없는 경우 각각 최대 5994개 및 12282개 뉴클레오티드의 시퀀스를 취할 수 있습니다.
이 저장소에 제시된 모델 컬렉션은 Instadeep의 허깅페이스 공간 nucleotide transformer 공간 및 Agro nucleotide transformer 공간)에서 사용할 수 있습니다!
흥미로운 연구 방향을 식별하는 데 도움이 된 건설적인 토론에 대해 Maša Roller와 Rostlab 회원, 특히 Tobias Olenyi, Ivan Koludarov 및 Burkhard Rost에게 감사드립니다. 또한 우리는 공공 데이터베이스에 실험 데이터를 보관하는 모든 사람, 이러한 데이터베이스를 유지 관리하는 사람, 분석 및 예측 방법을 무료로 제공하는 사람에게 감사를 표합니다. Jax 개발팀에도 감사드립니다.
이 저장소가 귀하의 작업에 유용하다고 생각되면 관련 논문 중 하나에 관련 인용을 추가하십시오.
nucleotide transformer 종이:
@article { dalla2023nucleotide ,
title = { The nucleotide transformer : Building and Evaluating Robust Foundation Models for Human Genomics } ,
author = { Dalla-Torre, Hugo and Gonzalez, Liam and Mendoza Revilla, Javier and Lopez Carranza, Nicolas and Henryk Grywaczewski, Adam and Oteri, Francesco and Dallago, Christian and Trop, Evan and Sirelkhatim, Hassan and Richard, Guillaume and others } ,
journal = { bioRxiv } ,
pages = { 2023--01 } ,
year = { 2023 } ,
publisher = { Cold Spring Harbor Laboratory }
}농업 nucleotide transformer 종이:
@article { mendoza2024foundational ,
title = { A foundational large language model for edible plant genomes } ,
author = { Mendoza-Revilla, Javier and Trop, Evan and Gonzalez, Liam and Roller, Ma{v{s}}a and Dalla-Torre, Hugo and de Almeida, Bernardo P and Richard, Guillaume and Caton, Jonathan and Lopez Carranza, Nicolas and Skwark, Marcin and others } ,
journal = { Communications Biology } ,
volume = { 7 } ,
number = { 1 } ,
pages = { 835 } ,
year = { 2024 } ,
publisher = { Nature Publishing Group UK London }
}세그먼트NT 용지
@article { de2024segmentnt ,
title = { SegmentNT: annotating the genome at single-nucleotide resolution with DNA foundation models } ,
author = { de Almeida, Bernardo P and Dalla-Torre, Hugo and Richard, Guillaume and Blum, Christopher and Hexemer, Lorenz and Gelard, Maxence and Pandey, Priyanka and Laurent, Stefan and Laterre, Alexandre and Lang, Maren and others } ,
journal = { bioRxiv } ,
pages = { 2024--03 } ,
year = { 2024 } ,
publisher = { Cold Spring Harbor Laboratory }
}코드와 모델에 대해 질문이나 피드백이 있으면 언제든지 저희에게 연락해 주세요.
우리 작업에 관심을 가져주셔서 감사합니다!


